
Resumo
Sitosterol, stigmasterol, betulinic acid, lupeol, 3-O-beta-D-glucopiranosylsitosterol, 3-O-alpha-L-rhamnopiranosylchromone, 5,7-dihydroxy-4'-methoxyisoflavone, 3',5,7-trihydroxy-4'-methoxyisoflavone and a mixture of two rel-2R,3S-3-O-alpha-L-rhamnopiranosilflavanonols were isolated from the roots of Andira fraxinifolia. Their structures were established by spectral data analysis.
Andira fraxinifolia; triterpenes; phenolic compounds
Andira fraxinifolia; triterpenes; phenolic compounds
ARTIGO
Constituintes fenólicos e terpenóides isolados das raízes de Andira fraxinifolia (Fabaceae)
Phenolic constituents and terpenoids isolated from the roots of Andira fraxinifolia (Fabaceae)
Virginia Claudia da SilvaI; Aline Nogueira AlvesI; Alessandra de SantanaI; Mário Geraldo de CarvalhoI, * * e-mail: mgeraldo@ufrrj.br ; Sandra Lúcia da Cunha e SilvaII; Jan SchripsemaIII
IDepartamento de Química, Instituto de Ciências Exatas, Universidade Federal Rural do Rio de Janeiro, Antiga Rodovia Rio-SP, km 47, 23851-900 Seropédica - RJ, Brasil
IIDepartamento de Estudos Básicos e Instrumentais, Campus de Itapetinga, Universidade Estadual do Sudoeste da Bahia, BA, Brasil
IIISetor de Química de Produtos Naturais, Centro de Ciência e Tecnologia, Universidade Estadual do Norte Fluminense, 28013-600 Campos dos Goytacazes - RJ, Brasil
ABSTRACT
Sitosterol, stigmasterol, betulinic acid, lupeol, 3-O-b-D-glucopiranosylsitosterol, 3-O-a-L-rhamnopiranosylchromone, 5,7-dihydroxy-4'-methoxyisoflavone, 3',5,7-trihydroxy-4'-methoxyisoflavone and a mixture of two rel-2R,3S-3-O-a-L-rhamnopiranosilflavanonols were isolated from the roots of Andira fraxinifolia. Their structures were established by spectral data analysis.
Keywords:Andira fraxinifolia; triterpenes; phenolic compounds.
INTRODUÇÃO
O gênero Andira compreende o grupo de vegetais popularmente conhecido por angelins, sendo representado por mais de 30 espécies1,2 distribuídas pela América Tropical e uma espécie na África2, sendo que a maioria é originária do Brasil1. No Brasil, foram encontradas 27 espécies e 7 variedades, sendo que o maior número de espécies se encontra na Amazônia1. Em face das propriedades vermífugas, esse gênero foi utilizado na Europa desde 1755, por médicos e farmacêuticos de diversos países que preconizavam a industrialização das cascas, transformando-as em pó, com o qual procuravam obter uma droga de aplicação anti-helmíntica1. Algumas espécies que pertencem ao gênero Andira ainda têm sido utilizadas popularmente como anti-helmínticas, apesar de seus efeitos tóxicos serem citados por diversos autores3-5. Em trabalho anterior divulgamos a avaliação da ação anti-helmíntica dos extratos brutos de A. anthelmia e A. fraxinifolia, sugerindo o extrato bruto da espécie A. anthelmia como um potente anti-helmíntico, porém necessitando de estudos complementares em virtude dos efeitos tóxicos detectados6. Trabalhos sobre o estudo químico deste gênero relatam principalmente a presença de isoflavonas7-12, flavanóis9,12, rotenóides13, compostos do tipo 2-arilbenzofurano-3-carbaldeídos12 e 2-aril-3-hidroximetil-benzofuranos13. Neste trabalho registra-se o resultado do primeiro estudo fitoquímico de A. fraxinifolia, descrevendo o isolamento e a identificação dos esteróides sitosterol (1) e estigmasterol (2), do 3-O-b-D-glicopiranosilsitosterol (1a), dos triterpenos lupeol (3), ácido betulínico (4), duas isoflavonas, biochanina A (5) e pratenseína (6), uma cromona, eucrifina (7) e dois 3-O-a-L-ramnopiranosil-flavanonóis (8 e 9).
Este é o primeiro registro das substâncias 1a, 3, 4, 7, 8 e 9 neste gênero. A ocorrência de isoflavonas tem sido relatada em todas as espécies de Andira já estudadas7-12, merecendo destaque a abundância da biochanina A. Podem ser utilizadas como marcadores taxonômicos deste gênero em Fabaceae.
RESULTADOS E DISCUSSÃO
O extrato metanólico das raízes de A. fraxinifolia foi submetido à partição com solventes. A fração obtida com hexano foi submetida ao fracionamento com técnicas cromatográficas e forneceu a mistura dos esteróides, sitosterol (1) + estigmasterol (2), lupeol (3) e ácido betulínico (4). Este mesmo tratamento da fração acetato de etila forneceu 3-O-b-D-glicopiranosilsitosterol (1a), 5,7-diidroxi-4'-metoxiisoflavona (5), 5,7,3'-triidroxi-4'-metoxiisoflavona (6), 5,7-diidroxi-3-O-a-L-ramnopiranosilcromona (7) e dois flavanonóis glicosilados, rel-2R,3S-5,7,4'-triidroxi-3-O-a-L-ramnopiranosilflavanonol (8) e rel-2R,3S-5,7,3',4'-tetraidroxi-3-O-a-L- ramnopiranosilflavanonol (9).
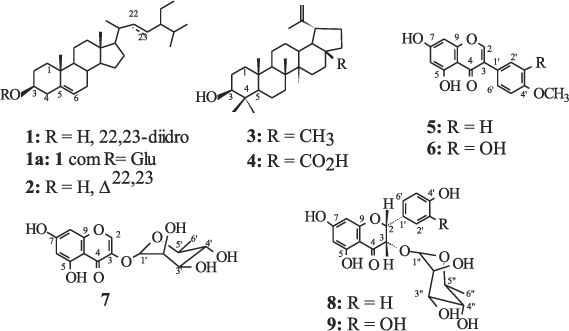
As estruturas do sitosterol (1) e estigmasterol (2)14, lupeol (3)15, do ácido betulínico (4)16 e do 3-O-b-D-glicopiranosilsitosterol (1a)17 foram identificadas através da análise dos espectros de RMN de 1H, 13C (BBD e DEPT-135 e 90), comparação com padrões usando CCDA e comparação com dados da literatura14-17.
As estruturas dos flavonóides 5 e 6 foram definidas através da análise dos espectros de RMN de 1H (500 MHz) e 13C (125 MHz) de cada um e comparação com dados na literatura18,19. Ambas apresentaram sinais em campo baixo [dH 12,91 (5) e 12,95 (6)] referentes ao grupo hidroxila formando ligação de hidrogênio com a carbonila, dois dubletos (J = 2Hz) representando os hidrogênios H-6 e H-8 [dH 6,20 e 6,37 (5); 6,18 e 6,33 (6)] e os singletos em dH 8,34 (5) e 8,29 (6) do H-2, que são compatíveis com o esqueleto de uma isoflavona. O espectro da substância 5 apresentou dois dubletos em dH 6,97 e 7,48 (8,7 Hz, 2H) de um sistema AA'BB' (H-2',6' e H-3',5') e o espectro de 6 apresentou três sinais em dH 6,94 (dl, 8,5 Hz, 1H), 6,97(d, 8,5 Hz, 1H) e 7,03 (sl, 1H) de um sistema ABC do anel B trissubstituído. Ambas apresentaram um singleto de grupo metoxila em dH 3,77 (5) e 3,79 (6). A localização desses grupos foi definida através da análise dos sinais de NOE nos espectros obtidos com experimentos NOEDIFF (200 MHz). O espectro resultante deste experimento com a substância 5, através de irradiação na freqüência da metoxila, apresentou sinal de NOE em dH 6,97 (H-3',5') e com 6 gerou NOE em dH 6,97 (d, H-5'), justificando a localização das metoxilas no carbono 4' de 5 e 6. Os espectros de massas dessas isoflavonas apresentaram, além dos picos em m/z 284 (M+. de 5) e 300 (M+. de 6), os picos em m/z 132 (25%, C9H8O, 5) e m/z 124 (10%, C7H8O2, 6) que justificam as estruturas propostas e, inclusive, a localização das metoxilas no anel B. Essas informações, a análise dos espectros de RMN de 13C e comparação com valores da literatura18,19 permitiram determinar as estruturas de 5 e 6 como 5,7-diidroxi-4'-metoxiisoflavona e 5,7,3'-triidroxi-4'-metoxiisoflavona, respectivamente.
A cromona (7) foi identificada como sendo a 5,7-diidroxi-3-O-a-L-ramnopiranosilcromona através da análise dos espectros de RMN de 1H e 13C (BBD e DEPT) e comparação com dados da literatura20. O espectro de RMN de 1H de 7 mostrou dois sinais duplos (J = 2,2 Hz) em dH 6,18 (1H); 6,35 (1H), um sinal simples de hidrogênio desprotegido em dH 8,22 (s, 1H). Esses dados e os valores dos deslocamentos químicos de carbonos em d 177,0 (C=O), 161,5 (C), 165,1 (C), 148,3 (CH), 138,0 (C), detectados nos espectros de RMN de 13C, permitiram propor uma unidade 5,7-diidroxicromona sustentando um substituinte em 3. Esses valores foram semelhantes aos descritos na literatura para esta unidade20. O dubleto em dH 5,22 (J = 1,4 Hz, H-1'), singleto largo em dH 3,89, multipletos em dH 3,58 e 3,25 e o dubleto em dH 1,10 (J = 6,0 Hz) permitram identificar a ramnose ligada na cromona. O espectro de RMN de 13C apresentou entre outros sinais de CH (71,5, 70,2, 69,9, 69,8), os sinais em d 17,8 (CH3) e 100,6 que foram atribuídos aos carbonos 6' e o sinal de 1' da ramnose. A localização da ramnose no C-3 da cromona foi confirmada pelo experimento de NOEDIFF cuja irradiação na freqüência do H-1' gerou NOE no hidrogênio em dH 8,22 (H-2). O espectro de massas de 7 não revelou o pico íon molecular, mas a presença do pico em m/z 194 (M+. C6H10O4) serviu para confirmar a proposta da cromona. Os deslocamentos químicos dos carbonos e hidrogênios de 7 foram semelhantes aos da literatura20, confirmando a estrutura da 5,7-diidroxi-3-O-a-L-ramnopiranosilcromona.
A análise dos espectros de RMN de 1H e 13C, incluindo técnicas especiais 1D e 2D, da subfração contendo flavanonóis e comparação com dados da literatura21-23 permitiram identificar a mistura dos glicosídeos 8 e 9. Os espectros de RMN de 1H, incluindo experimento 1H-1H-COSY desta mistura, apresentaram dois pares de dubletos acoplando entre si em dH 5,62/4,0 (J = 2,0 Hz), dH 5,55/4,21 (J = 2,0 Hz), representando dois sistemas AB em carbonos oxigenados, e um conjunto de sinais em dH 5,97-5,92 (contendo dubletos com J = 2,2 Hz) compatíveis com sinais de hidrogênios (H-6 e H-8) de flavanonas. A análise do espectro HMQC permitiu identificar os respectivos carbonos metínicos ligados aos hidrogênios representados por esses sinais e confirmar a natureza dos carbonos oxigenados-sp3 (dCH 80,1/73,1 e 79,9/73,4) e os carbonos não oxigendos-sp2 (95,2/96,2 e 95,2/96,2) que foram atribuídos aos respectivos carbonos C-2/C-3 (anel C) e C-6/C-8 (anel A) dos flavanonóis representados como 8 e 9, respectivamente. A proposta de mistura dos flavanonóis foi confirmada pela integração dos sinais dos respectivos pares de dubletos (J = 2,0 Hz), sendo que os sinais atribuídos a 8 [dH 5,62 (H-2) e 4,0 (H-3)] apresentaram-se com maior intensidade do que os atribuídos a 9 [(dH 5,55 (H-2) e 4,21 (H-3)]. A integração desses sinais permitiu calcular a percentagem relativa de 6/4 entre 8 e 9. Os valores das constantes de acoplamento (J = 2,0 Hz) são compatíveis com a relação cis entre os hidrogênios ligados aos carbonos metínicos C-2 e C-321. A análise dos espectros de RMN de 13C (BBD e APT) confirmou os deslocamentos dos carbonos metínicos e permitiu identificar os sinais dos carbonos quaternários desta unidade proposta para 8 e 9 [dC 192,9, 193,1(C=O), 164,1, 164,0 (C-5), 167,1, 167,1 (C-7), 162,6, 162,5 (C-9) e 100,2, 100,3 (C-10)]. Esses valores, aliados aos sinais de hidroxilas fenólicas em dH 11,7 (s, HO-5), 9,47 (sl), 8,92 (s) e 8,91 (s) estão de acordo com os anéis A e C desses flavanonóis. Os demais sinais na região de núcleos em sistemas aromáticos, presentes nos espectros de RMN de 1H, 13C e HMQC foram compatíveis com a presença do sistema AA'BB' [representado pelos sinais em dH 7,26 e 6,76 (d, 8,4 Hz, 2H, cada) ligados aos carbonos em dCH 127,8 e 115,1, respectivamente] e de um sistema ABC [representado pelos sinais em dH 6,84 (s, 1H), 6,76 (d, 8,4 Hz) e 6,71 (d, 8,4 Hz) ligados nos carbonos dCH 114,1, 117,6 e 115,1, respectivamente]. Os demais sinais de carbonos aromáticos quaternários [dC 125,8 (C-1', 8), 126,4 (C-1', 9), 157,3 (4', 8), 145,0, 145,2 (C-3' e C-4', 9)] estão de acordo com os padrões de substituição dos anéis B desses flavononóis. A unidade de açúcar foi identificada pelos pares de sinais em dCH 98,3/98,6, 70,3/70,3, 70,3/70,3, 71,3/71,2, 69,0/68,9 e dCH3 17,6/17,6 no espectro de RMN de 13C (BBD e APT) e pelos sinais de 1JCH presentes no espectro HMQC, que permitiu detectar os deslocamentos químicos dos hidrogênios [dH 4,74(s)/4,74(s), 3,44(m)/3,44(m), 3,17(m)/3,17(m), 3,03(m)/3,03(m), 2,44(m)/2,30(m), 0,83(d, 6,2 Hz)/0,79(d, 6,2 Hz)] ligados aos respectivos carbonos cujos dC são compatíveis com a ramnose. Os sinais de 2JCH e 3JCH no espectro HMBC da mistura permitiu verificar interações de acoplamento dos hidrogênios H-2',6' e H-3 com o C-2 (80,1), H-1" com C-3 (73,4) e H-3 com C-1" (98,8) e do H-2 com C-2',6'(127,8), que foram atribuídos a 8; e os sinais de acoplamento de H-3, H-6' e H-2' com C-2 (79,9), H-2 e H-1" com C-3 (73,3) e H-3 com C-1" (98,6), que foram atribuídos a 9. Estas informações permitiram propor as estruturas do rel-2R,3S-5,7,4'-triidroxi-3-O-a-L-ramnosilflavanonol (8) e rel-2R,3S-5,7,3',4'-tetraidroxi-3-O-a-L-ramnosilflavanonol (9). Os dados de RMN de 1H e 13C foram semelhantes aos registrados na literatura para os respectivos diastereoisômeros de 8 e 9, engeletina e astilbina22,23.
PARTE EXPERIMENTAL
Procedimentos experimentais
Os pontos de fusão foram determinados em placa de aquecimento MEL-TEMP II, Laboratory Devices USA, utilizando capilar, sem correção dos valores. Os espectros de massas de baixa resolução foram obtidos em cromatógrafo em fase gasosa acoplado à espectrometria de massas (CG-EM), modelo Saturn 2000 da Varian, utilizando sistema de "íon trap" e ionização por impacto de elétrons a 70 eV. Os espectros de Ressonância Magnética Nuclear, 1H e 13C (incluindo experimentos em 2D) foram registrados em espectrômetros Bruker DRX-500 (1H: 500 MHz e 13C: 125 MHz); Bruker Ac-200 (1H: 200 MHz e 13C: 50 MHz) e JEOL JNM-GX-400 (1H: 400 MHz e 13C: 100 MHz), utilizando CDCl3, Piridina-D5 ou DMSO-D6 como solventes e TMN como referência interna. Nas separações cromatográficas em coluna aberta usou-se sílica gel (Merck e Aldrich) ou Sephadex LH-20 (Aldrich); nas análises com camada fina utilizou-se sílica gel (Merck e Aldrich) com granulação adequada e revelação com luz UV (254 e 366 nm), AlCl3-EtOH (1%), reagente de Liebermann-Burchard e exposição em vapor de iodo.
Material vegetal
As raízes de Andira fraxinifolia foram coletadas em dezembro de 2001 na Reserva Biológica de Poço das Antas (RJ). Uma exsicata (nº 4617) encontra-se catalogada no Herbário RBR-UFRRJ, Departamento de Botânica, IB-UFRuralRJ.
Extração e isolamento
Após secagem e trituração, 630,0 g de raízes de A. fraxinifolia foram submetidas à extração através de maceração contínua com MeOH à temperatura ambiente. O extrato metanólico (AFRM, 92,0 g) foi submetido à partição com solventes em polaridades crescentes obtendo-se as frações com hexano (AFRMH, 4,6 g), AcOEt (AFRMA, 20,7 g) e MeOH (AFRMM, 43,5 g). A fração AFRMH foi cromatografada em coluna de sílica gel e eluída com hexano, mistura com CH2Cl2 e MeOH até 100% de MeOH, obtendo-se 44 subfrações de 15 mL. As subfrações 4-8 forneceram um precipitado cuja cristalização em MeOH forneceu uma mistura de 1 + 2 (60,0 mg). O grupo de subfrações 15-26 e 31-40 forneceu precipitados correspondentes, respectivamente, aos triterpenos 3 (79,0 mg, p.f. 210-212 0C, lit.24 212-215 0C) e 4 (66,0 mg, p.f. 283-285 0C, lit.24 282-285 0C). A fração AFRMA foi cromatografada em coluna de sílica gel e eluída com hexano, AcOEt e MeOH até 100% MeOH, obtendo-se 100 subfrações de 15 mL. As subfrações 43-100 (8,7 g) foram reunidas por análise em CCDA e submetidas à cromatografia em coluna de sílica gel, eluídas com AcOEt e mistura com MeOH até 100% MeOH, obtendo-se 42 subfrações de 50 mL. As subfrações 13-16 forneceram um precipitado em forma de placas, que foi identificado como a substância 1a (50,0 mg, p.f. 290-292 0C, lit.14 290-291 0C). As subfrações 17-22 após filtração em Sephadex LH-20 (MeOH 100%) forneceram a substância 5 (303,0 mg, p.f. 210-212 0C, lit.8 214-216 0C).As frações 23-25 (26,0 mg) continham uma mistura das substâncias 5 + 6. Esta fração foi purificada por meio de CCDP eluída com CHCl3/MeOH (9:1), dando origem à substância 6 (4,5 mg, p.f. 258-260 0C, lit.25 272-273 0C). As subfrações 27-30 (11,5 mg) foram reunidas e submetidas à coluna flash eluída com CHCl3/MeOH (8:2) obtendo-se cristais incolores em forma de agulhas, correspondentes à substância 7 (7,0 mg, p.f. 198-200 0C, lit.20 212-215 0C). As subfrações 32-36 (31,1 mg) foram reunidas e submetidas à coluna flash usando como eluente CHCl3/MeOH (8:2) onde foi possível obter um precipitado branco correspondente à mistura de 8 + 9 (14,0 mg).
rel-2R,3S-5,7,4'-triidroxi-3O-aL-ramnopiranosilflavanonol (8): RMN de 1H (400 MHz, DMSO-D6) dH (mult; J em Hz, H): 5,62 (d; 2,0; H-2), 4,0 (d; 2,0; H-3), 5,95 (d; 2,2; H-6), 5,97 (d; 2,2; H-8), 7,26 (d; 8,4; H-2',6'), 6,76 (d; 8,4; H-3',5'), 4,74 (s; H-1"), 3,44 (m; H-2"), 3,17 (m; H-3"), 3,03 (m; H-4") 2,44 (m; H-5"), 0,83 (d; 6,2; H-6"). RMN de 13C (100 MHz, DMSO-D6), dC: 80,1 (C-2), 73,1 (C-3), 192,9 (C-4), 164,1 (C-5), 95,2 (C-6), 167,1 (C-7), 96,2 (C-8), 162,6 (C-9), 100,2 (C-10), 125,8 (C-1'), 127,8 (C-2'), 115,1 (C-3'), 157,3 (C-4'), 115,1 (C-5'), 127,8 (C-6'), 98,3 (C-1"), 70,3 (C-2"), 70,3 (C-3"), 71,3 (C-4"), 69,0 (C-5"), 17,6 (C-6"). rel-2R,3S-5,7,3',4'-tetraidroxi-3-O-a-L- ramnopiranosilflavanonol (9): RMN de 1H (400 MHz, DMSO-D6) dH (mult; J em Hz; H): 5,55 (d; 2,0; H-2), 4,21 (d; 2,0; H-3), 5,92 (d;2,2; H-6), 5,94 (d; 2,2; H-8), 6,84 (s, H-2'), 6,71 (d, 8,4; H-5') , 6,76 (d; 8,4; H-6'), 4,74 (s; H-1"), 3,44 (m; H-2"), 3,17 (m; H-3"), 3,03 (m; H-4"), 2,30 (m; H-5"), 0,79 (d; 6,2; H-6"). RMN de 13C (100 MHz, DMSO-D6), dC: 79,9 (C-2), 73,4 (C-3), 193,1 (C-4), 164,0 (C-5), 95,2 (C-6), 167,1 (C-7), 96,2 (C-8), 162,5 (C-9), 100,3 (C-10), 126,4 (C-1'), 114,1 (C-2'), 145,0 (C-3'), 145,2 (C-4'), 115,1 (C-5'), 117,6 (C-6'), 98,6 (C-1"), 70,3 (C-2"), 70,3 (C-3"), 71,2 (C-4"), 68,9 (C-5"), 17,6 (C-6").
AGRADECIMENTOS
Ao CNPq, CAPES e à FAPERJ pelas bolsas concedidas e pelo apoio financeiro para o desenvolvimento dessa pesquisa.
Recebido em 2/8/05; aceito em 17/2/06; publicado na web em 6/7/06
- 1. Matos, N. F.; Acta Amazon 1979, 9, 241.
- 2. Pennington, T.; Lima, H. C.; Kew. Bull. 1995, 50, 557.
- 3. Rizzini, C. T.; Acad. Brasil. Ciências 1970, 42 (suplemento), 329.
- 4. Lapa-Bautista, A. R. P.; Lacaz, M. H. A.; Mors, W. B.; B.I.B.B 1976, 15, 31.
- 5. Pio Corrêa, M.; Dicionário de Plantas Úteis do Brasil e das Exóticas Cultivadas, Ministério da Agricultura-IBDF, Imprensa Nacional, Rio de Janeiro, 1984, vol. 1.
- 6. Silva, S. L. da C.; Borba, H. R.; do Bonfim, R. C. B.; de Carvalho, M. G.; Cavalcanti, H. L.; Barbosa, C.G.; Parasitol. Latinoam 2003, 58, 23.
- 7. Braz-Filho, R.; Gottlieb, O. R.; Pinho, S. L. V.; Monte, F. J. Q.; da Rocha, A.I.; Phytochemistry 1973, 12, 1184.
- 8. Lapa-Bautista, A. R. P.; Mors, W. B.; B.I.B.B. 1975, 14, 58.
- 9. Lock de Ugaz, O.; Sanchez, J. C. L.; Sanchez, R. P. U.; Tempesta, M. S.; Fitoterapia 1991, 62, 89.
- 10. Kraft, C.; Jenett-Siems, K.; Siems, K.; Gupta, M. P.; Bienzle, U.; Eich, E.; J. Ethnopharmacol 2000, 73, 131.
- 11. da Silva, B. P.; Velozo, L. S. M.; Parente, J. P.; Fitoterapia 2000, 71, 663.
- 12. Kraft, C.; Jenett-Siems, K.; Siems, K.; Solis, P. N.; Gupta, M. P.; Bienzle, U.; Eich, E.; Phytochemistry 2001, 58, 769.
- 13. Kraft, C.; Jenett-Siems, K.; Köhler, I.; Siems, K.; Abbiw, D.; Bienzle, U.; Eich, E.; Z. Naturforsch., C: J. Biosci 2002, 57, 785.
- 14. Dutra, N. N.; Alves, H. de M.; de Carvalho, M. G.; Braz-Filho, R.; Quim. Nova 1992, 15, 10.
- 15. Sobrinho, D. C.; Hauptli, M. B.; Appolinário, E. V.; Kollenz, C. L. M.; de Carvalho, M. G.; Braz-Filho, R.; J. Braz. Chem. Soc 1991, 2, 15.
- 16. Bilia, R. A.; Mendez, J.; Morelli, I.; Pharmaceutica Acta Helvetiae 1996, 71, 191.
- 17. Kojima, H.; Sato, N.; Hatano, A.; Ogura, H.; Phytochemistry 1990, 29, 2351.
- 18. dos Santos, S. A.; de Carvalho, M. G.; Braz-Filho, R.; J. Braz. Chem. Soc, 1995, 6, 349.
- 19. Agrawal, P. K.; Bansal, M. C.; Porter, L. J.; Foo, L. Y. Em Carbon-13 NMR of Flavonoids - Studies in Organic Chemistry 39; Agrawal, P. K., ed.; Elsevier Science Publishers B. V.: Amsterdam, 1989, vol. 39, p.201.
- 20. Tschesche, R.; Delhvi, S.; Sepulveda, S.; Breitmaier, E.; Phytochemistry 1979, 18, 867.
- 21. Gaffield, W.; Waiss Jr, A. C.; J.Org. Chem. 1975, 40, 1057.
- 22. Silva, D. H. S.; Yoshida, M.; Kato, M. J.; Phytochemistry 1997, 46, 579.
- 23. Cintra, P.; Malaspina, O.; Petacci, F.; Fernandes, J. B.; Bueno, O. C.; Vieira, P. C.; da Silva, M. F. G.; J.Braz. Chem. Soc. 2002, 13, 115
- 24. Ahmad, V. U.; Rahman, A.; Handbook of Natural Products Data, Elsevier: Amsterdam, 1994, vol. 2.
- 25. Buckingham, J.; ed. Dictionary of Organic Compounds, Chapman and Hall: London, 1982, 5th e. (T-03375).
Datas de Publicação
-
Publicação nesta coleção
06 Set 2011 -
Data do Fascículo
Dez 2006
Histórico
-
Aceito
17 Fev 2006 -
Recebido
02 Ago 2005
