Resumo
The effect of substituents on the energies and geometries of 3-hydroxypropenal was studied using the B3LYP/6-311++G(d,p) model. The hydrogen bond energies indicate that the strongest donors and the weakest acceptors present the highest and the weakest hydrogen bonds, respectively, indicating the validity of the Madsen RAHB model. Geometric parameters indicate that the intensity of the hydrogen bond is proportional to the resonance, as suggested by the RHAB model. The effect of substituents diverges from the model proposed by Gilli et al. Sometimes the results indicate that the donor or acceptor effect is more important than the point of substitution.
3-hydroxypropenal; RAHB; effect of substituents
3-hydroxypropenal; RAHB; effect of substituents
ARTIGO
Efeitos de substituintes na ligação de hidrogênio do 3-hidroxipropenal
Effects of the substituents on the hydrogen bond of 3-hydroxypropenal
Valéria C. F. Rustici; Giovanni F. Caramori; Sérgio E. Galembeck* * e-mail: segalemb@usp.br
Departamento de Química, Faculdade de Filosofia Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Av. Bandeirantes, 3900, 14040-901 Ribeirão Preto - SP, Brasil
ABSTRACT
The effect of substituents on the energies and geometries of 3-hydroxypropenal was studied using the B3LYP/6-311++G(d,p) model. The hydrogen bond energies indicate that the strongest donors and the weakest acceptors present the highest and the weakest hydrogen bonds, respectively, indicating the validity of the Madsen RAHB model. Geometric parameters indicate that the intensity of the hydrogen bond is proportional to the resonance, as suggested by the RHAB model. The effect of substituents diverges from the model proposed by Gilli et al. Sometimes the results indicate that the donor or acceptor effect is more important than the point of substitution.
Keywords: 3-hydroxypropenal; RAHB; effect of substituents.
INTRODUÇÃO
Os compostos b-dicarbonílicos, e particularmente as b-dicetonas despertaram o interesse de muitos pesquisadores em função de suas variadas aplicações. Os complexos metálicos formados por b-dicetonas têm sido bastante empregados como aditivos para combustíveis, fluidos supercríticos, em inúmeras reações de condensação principalmente reações orgânicas1-3, na manufatura de filmes supercondutores, na produção de catalisadores homogêneos e heterogêneos4 e em química de polímeros5.
Os complexos lantanídicos de compostos b-dicarbonílicos são usados como marcadores luminescentes em imunologia (fluoroimunoanálise)6,7. Os b-dicetonatos de lantanídeos possuem amplas aplicações na separação de misturas de lantanídeos com outros metais por sublimação fracionada, em cromatografia gasosa, em cromatografia líquida de adsorção, em análises espectrofotométricas, como oxidantes e como reagentes de deslocamento em espectroscopia de RMN6,7.
Dentre estes compostos, um dos mais conhecidos é a 2,4-pentanodiona (acetilacetonato). Em estudos realizados por Verdenelli8, o acetilacetonato é empregado na síntese de dióxido de estanho que, posteriormente, é utilizado como condutor em células solares. Outros empregos, recentemente relatados, são na síntese de moléculas com propriedades farmacêuticas como analgésicos e antitérmicos9.
É bem conhecido que o tautômero mais estável do composto b-dicarbonílico 1,3-propanodial é, na forma cetoenólica, o 3-hidroxipropenal (1HB), caracterizado por um sistema tipo anel fechado assimétrico estabilizado por uma ligação de hidrogênio muito intensa, O-H×××O, intramolecular10 (Figura 1). Gilli e colaboradores11,12 propuseram o modelo de ligações de hidrogênio assistidas por ressonância, ("Resonance Assisted Hydrogen Bond", RAHB), para racionalizar o comportamento das ligações de hidrogênio O-H×××O e N-H×××O muito curtas em sistemas conjugados. Em uma primeira etapa desse modelo seqüencial, as cargas parciais existentes nos oxigênios da forma canônica dipolar levariam a uma atração entre esses dois átomos e, por conseqüência, a uma menor distância O×××O. Em uma segunda etapa, o átomo de hidrogênio positivo que participa da ligação de hidrogênio se move para o oxigênio com carga parcial negativa, levando a uma neutralização das cargas opostas, localizadas nos oxigênios, e a um incremento da delocalização eletrônica no sistema p, (Figura 1a). A ligação de hidrogênio muito curta e muito forte formada por esse mecanismo possui um caráter covalente apreciável e é uma ligação de três centros e quatro elétrons, semelhante ao modelo proposto por Pimentel13 para a ligação de hidrogênio no ânion [F×××H×××F]- . Madsen e co-autores14, através da determinação experimental de densidades eletrônicas na benzoilacetona, modificaram este modelo e propuseram que os oxigênios apresentassem cargas parciais negativas e o hidrogênio situado entre eles, carga positiva. O modelo RAHB foi generalizado e ampliado por Gilli e inserido no modelo de ligação de hidrogênio eletrostático-covalente ("Electrostatic Covalent H-Bond Model" (ECHBM))15.
A intensidade da ligação de hidrogênio intramolecular em derivados do 3-hidroxipropenal pode ser afetada pela presença de substituintes que, dependendo de sua natureza, dimensão e posição podem induzir diferentes efeitos na delocalização dos elétrons p15,16. Estas interações apresentam grande influência sobre a estrutura eletrônica desta classe de compostos, conferindo a estes propriedades específicas, as quais têm sido caracterizadas através de técnicas espectroscópicas como ressonância magnética nuclear e espectroscopia de infravermelho17. Gilli e colaboradores11,12 verificaram que as RAHB são estabilizadas por substituintes retiradores de elétrons no C(1) ou doadores de elétrons no C(3), pois assim seria gerada uma grande carga parcial positiva no oxigênio enólico, O(1), e uma grande carga parcial negativa no oxigênio carbonílico18, O(3), (Figura 1). Este modelo geral foi proposto a partir da comparação da estrutura cristalina de uma série de compostos contendo RAHB, como os enóis de b-dicetonas, b-cetoésteres e b-cetoamidas18, e assim deveria ser aplicável a quaisquer outros compostos que contenham RAHB. Apesar dos vários trabalhos que estudaram o efeito de substituintes no 3-hidroxipropenal13-18, nenhum deles estudou de maneira uniforme o efeito de substituintes doadores e retiradores de elétrons na estrutura e na ligação de hidrogênio desse composto. Assim, nesse trabalho investigamos o efeito de substituintes nas energias das ligações de hidrogênio, nos parâmetros geométricos e nas cargas do 3-hidroxipropenal.
PARTE EXPERIMENTAL
Métodos computacionais
Todos os cálculos foram realizados utilizando-se o programa Gaussian 9819. As otimizações de geometria e as determinações de freqüências vibracionais foram realizadas utilizando-se o método B3LYP20 e o conjunto de funções de base 6-311++G(d,p)21. Todos os compostos foram otimizados sem restrições, exceto 8HB, para o qual se manteve o comprimento de ligação O(1)-H(1) fixo em 0,970 Å, pois o H(1) tende a migrar espontaneamente para o O(3). Foram realizados testes com o método MP2 e a mesma base e os resultados foram muito similares aos obtidos com o método DFT.
As energias das ligações de hidrogênio, EHB-NHB, foram calculadas pela diferença entre as formas quelato, HB, e aberta, NHB, corrigidas pelas energias de ponto zero, utilizando-se fator de escalonamento 1,0000. Vários autores criticaram essa definição e buscaram formas alternativas para se obter essa grandeza, normalmente envolvendo sistemas modelo ou correlações com parâmetros geométricos ou obtidos pela análise topológica da densidade eletrônica17,22,23. Contudo, verificou-se que na ausência de interações secundárias na forma aberta, como ligações de hidrogênio ou repulsões estéricas, o valor de EHB-NHB calculado como a diferença entre as energias de HB e NHB produz resultados confiáveis17. As energias da ligação de hidrogênio também foram calculadas por um modelo mecânico quântico simples, o de um elétron em um anel, com massas efetivas diferentes para a ligação de hidrogênio e o sistema conjugado24. Esta energia foi denominada 2DE1.
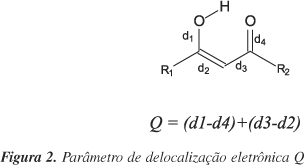
Na análise das geometrias foram empregados os parâmetros Dr25, Q12, <l>26 e RC26. O primeiro corresponde à diferença entre a soma do raio de van der Waals dos átomos de H e do oxigênio receptor e a distância H×××O. De acordo com critérios geométricos necessários para a ocorrência da ligação de hidrogênio25,26, essa distância deverá ser menor que a soma dos raios de van der Waals; assim, os maiores valores de Dr implicam em ligações de hidrogênio mais fortes. O parâmetro Q é usado para descrever a delocalização eletrônica (Figura 2), pois este é uma medida direta da equalização dos comprimentos das ligações equivalentes da estrutura central13. O parâmetro Q tem apresentado boa correlação com outros parâmetros que descrevem ligações de hidrogênio, como as distâncias O×××O e O×××H12. Através do modelo RAHB, quanto menor o valor de Q, mais intensa é a ligação de hidrogênio e maior a delocalização eletrônica12.
O índice médio de delocalização, <l>, é definido como:
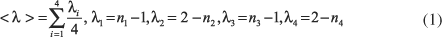
onde ni são as ordens de ligação propostas por Pauling27

e d(n) e d(1) são os comprimentos de ligação para as ordens de ligação n e 1, respectivamente e c é uma constante que depende do tipo de ligação química. Este índice é diretamente proporcional à equalização dos comprimentos de ligação, ou do incremento da ressonância. RC é a diferença entre d(H×××O) e d(O-H), ou seja, esse parâmetro é inversamente proporcional à extensão da transferência do próton do O(1) para o O(3), ou refletiria o enfraquecimento da ligação de hidrogênio26. As cargas atômicas foram calculadas pelo método "Natural Population Analysis (NPA)"28.
RESULTADOS E DISCUSSÃO
Energias das ligações de hidrogênio
Pela Tabela 1, pode-se observar que não há uma boa correlação entre as energias das ligações de hidrogênio calculadas pelos dois métodos, EHB-NHB e 2DE1. Isso pode indicar que os valores de 2DE1 não são suficientemente precisos ou que esse modelo necessita sofrer modificações. Por esta Tabela verifica-se que, para os substituintes localizados em R1, EHB-NHB varia segundo: 6 > 2 > 1 > 4 > 8 > 10, assim os doadores de elétrons mais fortes apresentarão ligações de hidrogênio mais intensas e os retiradores de elétrons as menores energias. Isso se encontra em contraste com as observações de Gilli e colaboradores18 e sugere que o O(1) pode apresentar uma carga parcial negativa, como observado por Madsen e colaboradores14 para a ordem de estabilidade dos substituintes em R1. A substituição em R2 apresenta um efeito semelhante ao observado para R1, indicando que a doação de densidade eletrônica desestabiliza o próton localizado no O(1), que corresponde ao modelo proposto por Gilli e colaboradores18.
Parâmetros geométricos
A Tabela 2 apresenta os parâmetros geométricos das ligações de hidrogênio intramoleculares para os derivados do 3-hidroxipropenal, dentre os quais, os valores das distâncias de ligação O-H (d(O-H)), O×××O (d(O×××O)) e H×××O (d(H×××O)), bem como os parâmetros Dr, Q, RC e <l>.
As correlações entre diferentes parâmetros geométricos, ou as correlações estruturais, foram utilizadas largamente por Gilli, na elaboração do modelo RAHB e em várias extensões29. Elas são de interesse particular pois podem indicar uma contribuição da ligação de hidrogênio na delocalização eletrônica pelo sistema conjugado. Correlações entre os vários parâmetros geométricos e a energia da ligação de hidrogênio, EHB-NHB, foram menos estudadas, mas Grabowski obteve uma boa correlação entre essa energia e o parâmetro Q para os derivados fluorados e clorados23. Procurou-se explorar as correlações entre EHB-NHB e os vários parâmetros geométricos apresentados na Tabela 2, mas em nenhum caso se obteve resultados satisfatórios.
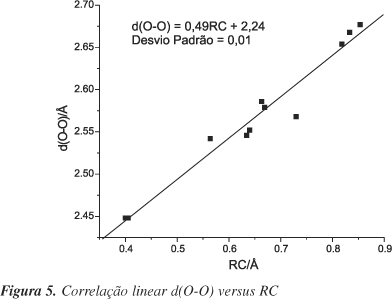
Pode-se notar que algumas das grandezas listadas na Tabela 2 apresentam boa correlação linear, como Dr e Q, com coeficiente de correlação, (r), = 0,929; RC e <l>, r = 0,930, (Figura 3); d(O×××O) e <l>, r = 0,910, (Figura 4); e d(O×××O) e RC, r = 0,980, (Figura 5). Correlações entre alguns desses parâmetros foram obtidas por Gilli e colaboradores26. Essas correlações indicam que o incremento da ligação de hidrogênio conduz a um aumento da ressonância, como proposto pelo modelo RAHB18. O fortalecimento da ligação de hidrogênio pode ser indicado por RC, ou d(O×××O), pois Gilli et al.18 observaram que a transferência parcial do próton para o oxigênio carbonílico ou a distância entre os oxigênios se correlacionam com o comprimento da ligação de hidrogênio.
De acordo com os parâmetros geométricos analisados, as substituições em R1 (vizinhas ao grupo enol) são as responsáveis pelas maiores variações nos parâmetros geométricos proporcionais à força da ligação de hidrogênio intramolecular e à delocalização dos elétrons p. É interessante notar que apesar dos substituintes apresentarem características diferentes, a influência de todos eles na força da ligação de hidrogênio intramolecular foi a mesma, indicando que os efeitos da posição da substituição estão prevalecendo sobre o caráter doador de elétrons ou receptor de elétrons dos mesmos. No entanto, as substituições em R2 apresentaram resultados diferentes dos obtidos pelas substituições em R1. A presença de substituintes retiradores de elétrons, Cl, F e NO2, diminui a ressonância no anel e conseqüentemente, observa-se uma diminuição na força de ligação de hidrogênio. A presença de CH3 leva a um ligeiro aumento na força da ligação de hidrogênio e da delocalização dos elétrons p, fornecendo resultados muito semelhantes aos observados para o 3-hidroxipropenal, pois este grupo é um fraco doador de elétrons. Como se pode observar, nesse caso os efeitos dos substituintes se sobressaem sobre os efeitos da posição da substituição. É interessante notar que esses dados se encontram de acordo com o modelo proposto por Gilli e colaboradores para o efeito de substituintes no 3-hidroxipropenal18. Contudo, os parâmetros apresentados na Tabela 2 não variam monotonicamente com a intensidade do caráter receptor e doador de elétrons para os substituintes localizados em R1 ou em R2. As exceções são d(H×××O) , d(O×××O) e Dr para substituintes em R2, excluindo-se o substituinte metila. Os dois primeiros parâmetros crescem e o terceiro decresce, de acordo com a ordem de caráter receptor: 7, 3, 1, 5, 9 e 11. O fato de não se ter observado correlação linear para as demais grandezas pode ser atribuído às pequenas variações observadas nos parâmetros geométricos e também pelo fato que alguns dos substituintes indicados apresentam caráter doador ou receptor de elétrons p e sigma.
As observações apresentadas acima não estão totalmente de acordo com o modelo proposto por Gilli et al.18 para os efeitos de substituintes. Um exemplo é o efeito dos substituintes em R2. Pelo modelo proposto pelo grupo de Ferrara esperar-se-ia que os substituintes doadores de elétrons tornassem a ligação de hidrogênio mais forte em R2, enquanto substituintes retiradores de elétrons a enfraquecessem. Isso indica que o modelo não prevê o papel dos substituintes doadores de elétrons.
As conclusões acima são reforçadas pela análise dos parâmetros listados na Tabela 2 para os compostos na conformação aberta, NHB. Os valores de Q são muito maiores e constantes, e os valores de <l> são menores que os da forma quelato, HB, indicando que as substituições não afetam ou afetam muito pouco a delocalização eletrônica na ausência de ligação de hidrogênio intramolecular.
Cargas atômicas
A Tabela 3 apresenta os valores das cargas NPA para todos os compostos estudados. Os valores entre parênteses correspondem à diferença de carga entre os compostos HB e NHB, D(HB-NHB). Um dado interessante é que todos os hidrogênios, das estruturas (HB), têm aproximadamente os mesmos valores de carga, em média 0,506 elétron, e são sempre maiores que as cargas dos H(1) das estruturas abertas, (NHB). Podemos destacar que os compostos 10HBe 8HBpossuem os maiores valores de carga positiva em H(1), (0,514 e 0,513 elétron), e 11HBo menor valor (0,484 elétron). Isto mostra que não apenas a posição de um dado substituinte, bem como sua natureza, pode provocar efeitos diferenciados na distribuição de carga da molécula. Apesar das cargas nos átomos de H(1), nos compostos HB, não apresentarem grandes variações, as cargas dos átomos de oxigênio variaram consideravelmente de HB para NHB.
Comparando as duas conformações, temos que a presença da ligação de hidrogênio atribui um aumento significativo de carga negativa nos oxigênios. O oxigênio da carbonila, O(3), é o átomo mais afetado pela presença da ligação de hidrogênio. Dentre os compostos substituídos em R1, 4HB, 10HBe 6HBapresentaram os maiores valores de Dq(HB-NHB) (-0,089 -0,091 e -0,094 elétron, respectivamente), indicando que a ligação de hidrogênio intramolecular promove um aumento da densidade eletrônica nas carbonilas. Para os demais, observaram-se valores com ordens de grandeza parecidas, por ex., -0,085 e -0,086 elétron para os compostos 8HBe 2HB. Para substituições em R2, a estrutura 5HBapresentou o maior valor de D(HB-NHB) (-0,086 elétron), seguida dos compostos 7HB, 3HBe 9HBque apresentaram valores muito próximos dos obtidos para os compostos com substituições em R1, -0,077, -0,082 e -0,083 elétron, respectivamente. Isto mostra que a presença de ligação de hidrogênio torna a delocalização eletrônica mais acentuada no interior do anel. Além disso, o aumento de carga negativa de O(3), para todos os substituintes, indica que as variações de carga na carbonila são pouco afetadas pela substituição, bem como pela natureza do substituinte.
Com relação aos oxigênios dos grupos hidroxila, observa-se, para as estruturas com substituição em R1 ou em R2, que a presença da ligação de hidrogênio também causou aumento de carga negativa em O(1). Este aumento não é tão intenso como no caso do grupo carbonila. A presença de substituintes tanto em R1 quanto em R2 provoca modificações muito variadas, como por ex., nos valores de variação de carga nos casos 6HB(-0,052 elétron) e 7HB (-0,026 elétron). Devemos destacar que em relação aos substituintes CH3, Cl e NH2, a substituição em R1 afeta mais a carga de O(1) que a substituição em R2. As cargas elevadas para O(1) e O(3) indicam a validade do modelo de Madsen et al14.
Com relação ao carbono C(1), observa-se que para os compostos com substituição em R1, os substituintes que provocam maiores variações em D(HB-NHB) são Cl e CH3, enquanto que as variações provocadas por NH2 e F são semelhantes. O mesmo se observa para os compostos substituídos em R2. Com relação ao carbono C(2), nota-se que para os compostos com substituição em R1, os grupos NH2 e F provocam diminuições no valor de D(HB-NHB). Já para os compostos com substituição em R2, não se observam grandes variações nos valores de D(HB-NHB) para C(2). Com relação ao carbono C(3), observa-se para os compostos com substituições em R1 que o grupo NH2 torna D(HB-NHB) mais negativo que os demais grupos; isso significa que a ligação de hidrogênio provoca uma diminuição da carga de C(3). No entanto, em relação aos compostos com substituição em R2, observa-se que os valores de D(HB-NHB) são muito parecidos.
A partir destas análises tentou-se fazer uma conexão entre os dados obtidos das cargas parciais e os resultados da análise dos parâmetros geométricos. De acordo com estudos teóricos30, a distribuição de carga nos átomos de oxigênio dos grupos carbonila e hidroxila pode ser afetada pela substituição, o que por razões eletrostáticas altera as distâncias O×××O, influenciando diretamente na força da ligação de hidrogênio. No entanto, tais propriedades não foram observadas para os compostos estudados quando se comparou o malonaldeído (1HB) e seus derivados.
Com relação aos compostos HB, as substituições em R1 provocaram um aumento das cargas negativas em O(3). No entanto, as cargas de O(1) sofreram poucas variações ou mantiveram-se constantes. Para aos compostos 4HBe 6HB, que apresentam ligações de hidrogênio mais intensas e maior deslocalização dos elétrons p, esperava-se uma diminuição das cargas negativas nos oxigênios O(1) e O(3), porém o que se observou foi um aumento das mesmas, quando comparadas às cargas de 1HB(Tabela 3), o que está em desacordo com os resultados de Trendafilova e colaboradores30. Os compostos que possuem oxigênios de carbonilas com pequena densidade eletrônica nem sempre são os que possuem ligações de hidrogênio estáveis. De acordo com os parâmetros geométricos, o composto 6HBapresenta a ligação de hidrogênio mais forte e maior delocalização eletrônica p, porém observa-se que a carga parcial de O(3) é mais negativa em 6HBque em 1HB(-0,619 elétron).
Devido aos resultados obtidos pela análise das cargas de O(1) e O(3), optou-se pela análise das variações de cargas entre os átomos O(1) e C(1), D(O(1)-C(1)) e entre O(3) e C(3), D(O(3)-C(3)) (Tabela 4). Com relação aos compostos substituídos em R1, observou-se que os valores de D(O(3)-C(3)) se mantiveram próximos, com exceção de 6HB. Por outro lado, os valores de D(O(1)-C(1)) diminuíram; isso mostra que a carga de C(1) se torna mais positiva para os compostos substituídos que em 1HB.
Os resultados obtidos justificam o fato de 6HBapresentar a ligação de hidrogênio mais intensa, pois o valor de D(O(1)-C(1)) sendo menos negativo que o de D(O(3)-C(3)) facilita a transferência de próton na ligação de hidrogênio. Os dados obtidos para as substituições em R2, mostram o inverso. Os valores de D(O(1)-C(1)) mostram-se equivalentes, enquanto que os valores de D(O(3)-C(3)) se tornam menos negativos em 3HB-9HB.
Algumas peculiaridades observadas confirmam os resultados das análises anteriores. Para os compostos com substituição em R2, observa-se que 3HBé o que apresenta maior força de ligação de hidrogênio e também a maior delocalização eletrônica. Isto pode ser observado pelo valor de D(O(3)-C(3)). Além disso, os compostos 8HBe 9HBapresentam valores positivos na variação de carga, o que mostra que as cargas positivas são muito acentuadas nos átomos C(1) e C(3). No primeiro caso, 8HB, o qual não é um mínimo de energia, a carga de C(1) é muito positiva e associada a um grande valor de D(O(3)- C(3)) (-0,247 elétron). Este fato pode ser um indício da causa da instabilidade dessa estrutura. Para 9HBtemos que D(O(1)-C(1)) é de -0,332, ou seja, muito semelhante a 1HB, porém o valor de D(O(3)-C(3)) é de +0,250 elétron, o que indica que a carga do C(3) é mais positiva em relação à carga de O(3). Isso justifica o fato da ligação de hidrogênio de 9HBser mais fraca.
CONCLUSÕES
Neste trabalho foram estudados as energias e os parâmetros geométricos de derivados do 1,3-hidroxipropenal substituídos por grupos doadores e retiradores de elétrons. As energias foram calculadas pelas diferenças entre as formas aberta NHB e fechada HB, bem como pelo modelo da partícula em um anel. Ambos não apresentaram boa correlação entre si. A energia calculada por esses modelos não apresenta correlação, o que nos leva a questionar a validade do último modelo. Por outro lado, as variações energéticas para os substituintes em C(1) e C(3) são semelhantes, em contraste com o modelo proposto por Gilli e colaoradores, sugerindo que a modificação do modelo RAHB proposta por Madsen e colaboradores é apropriada. Os parâmetros Dr e Q bem como <l> e RC são linearmente correlacionados, o que indica que um incremento da força da ligação de hidrogênio produz um incremento na ressonância do sistema insaturado, indicando a validade do modelo RAHB. Substituintes doadores ou retiradores de elétrons em R1 levam a um incremento na ressonância e na força da ligação de hidrogênio. Substituintes em R2 apresentam um efeito muito menor que os localizados em R1 e não apresentam qualquer padrão de comportamento. Observaram-se boas correlações entre d(O×××O) e <l> e d(O×××O) e RC. Contudo, a distribuição dos pontos nos gráficos não é totalmente compatível com o modelo proposto por Gilli e colaboradores. Os dados obtidos permitem que se conclua que o modelo proposto por esses autores para os efeitos de substituintes em compostos que apresentam RAHB apresenta discrepâncias e que os efeitos de substituintes são mais importantes que o sítio de substituição. As análises das cargas atômicas obtidas pelo método NPA confirmam as observações obtidas a partir dos parâmetros geométricos.
AGRADECIMENTOS
À FAPESP, CAPES e ao CNPq pelo financiamento desse projeto e ao LCCA-USP pela generosa alocação de recursos computacionais. S. E. Galembeck agradece ao CNPq pela bolsa de produtividade em pesquisa. V. C. F. Rustici e G. F. Caramori agradecem à FAPESP pelas bolsas de doutorado.
REFERÊNCIAS
1. Zhu, S.; Xu, B.; Zhang, J.; J. Fluorine Chem. 1995, 74, 167.
2. Kruger, P. E.; Moubaraki, B.; Murray, K. S.; Polyhedron 1997, 16, 2659.
3. Rodrigues, F.; Canac, Y.; Lubineau, A.; Chem. Commun. 2000, 20, 2049.
4. Eng, S. J.; Motekaitis, R. J.; Martell, A. E.; Inorg. Chim. Acta 1998, 278, 170.
5. David, L.; Craciun, C.; Cozar, O.; Chis, V.; Agut, C.; Rusu, D.; Rusu, M.; J. Mol. Struct. 2001, 563, 573
6. Sabatini, N.; Guardigli, M.; Lenh, J. M.; Coord. Chem. Rev. 1993, 123, 201.
7. Alexander, V.; Chem. Rev. 1995, 95, 273.
8. Verdenelli, M.; Parola, S.; Hubert-Pfalzgraf, L. G.; Lecocq, S.; Polyhedron 2000, 19, 2069.
9. Arias, J. L.; Cabrera, A.; Sharma, P.; Rosas, N.; Garcia, J. L.; Hernandes, S.; Inorg. Chim. Acta 2000, 310, 261.
10. Buemi, G.; Zuccarello, F.; Chem. Phys. 2004, 306, 115.
11. Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V.; J. Am. Chem. Soc. 1989, 111, 1023.
12. Gilli, G.; Bertolasi, V.; Ferretti, V.; Gilli, G.; J. Am. Chem. Soc. 1994, 116, 909.
13. Pimentel, G. C.; McClellan, A. L.; The Hydrogen Bond, 1st ed, W. H. Freeman and Company, Reinhold Publishing Corporation: New York, 1960.
14. Madsen, G. K. H.; Iversen, B. B.; Larsen, F. K.; Kapon, M.; Reisner, G. M.; Herbstein, F. H.; J. Am. Chem. Soc. 1998, 120, 10040.
15. Gilli, G.; Gilli, P.; J. Mol. Struct. 2000, 552, 1.
16. Rios, M. A.; Rodriguez, J.; J. Mol. Struct. (Theochem) 1991, 228, 149.
17. Buemi, G.; Zuccarello, F.; J. Mol. Struct. (Theochem) 2002, 581, 71.
18. Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G.; J. Am. Chem. Soc. 1991, 113, 4917.
19. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E. Jr.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Radbuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, L.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T. A.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A.; Gaussian 98, Gaussian Inc.: Pittsburgh PA, 1998.
20. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 37, 785.
21. Foresman, J. B.; Frisch, E.; Exploring Chemistry with Electronic Structure Methods, 2nd ed., Gaussian, Inc.: Pittsburgh, 1996.
22. Cuma, M.; Scheiner, S.; Kar, T.; J. Mol. Struct (Theochem) 1999, 467, 37.
23. Grabowski, S. J.; J. Mol. Struct. 2001, 562, 137.
24. Munn, R.W.; Eckhardt, C. J.; J. Phys. Chem. A 2001, 105, 6938.
25. Grabowski, S. J.; J. Phys. Org. Chem. 2004, 17, 18.
26. Gilli, P.; Bertolasi, V.; Pretto, L.; Lycka, A.; Gilli, G.; J. Am. Chem. Soc. 2002, 124, 13554.
27. Pauling, L.; J. Am. Chem. Soc. 1947, 69, 542; Pauling, L.; The Nature of the Chemical Bond, 3rd ed., Cornell University Press: Ithaca, NY, 1960. ; Bürgi, H.-B.; Dunitz, J. D.; J. Am. Chem. Soc. 1987, 109, 2924.
28. Reed, A. E.; Weinstock, R. B.; Weinhold, F.; Chem. Phys. 1985, 83, 735.
29. Gilli, P.; Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, G.; J. Am. Chem. Soc. 2004, 126, 3845.
30. Trendafilova, N.; Bauer, G.; Mihaylov, T.; Chem. Phys. 2004, 302, 95.
Recebido em 2/8/05; aceito em 21/2/06; publicado na web em 13/7/06
- 1. Zhu, S.; Xu, B.; Zhang, J.; J. Fluorine Chem. 1995, 74, 167.
- 2. Kruger, P. E.; Moubaraki, B.; Murray, K. S.; Polyhedron 1997, 16, 2659.
- 3. Rodrigues, F.; Canac, Y.; Lubineau, A.; Chem. Commun 2000, 20, 2049.
- 4. Eng, S. J.; Motekaitis, R. J.; Martell, A. E.; Inorg. Chim. Acta 1998, 278, 170.
- 5. David, L.; Craciun, C.; Cozar, O.; Chis, V.; Agut, C.; Rusu, D.; Rusu, M.; J. Mol. Struct. 2001, 563, 573
- 6. Sabatini, N.; Guardigli, M.; Lenh, J. M.; Coord. Chem. Rev 1993, 123, 201.
- 7. Alexander, V.; Chem. Rev 1995, 95, 273.
- 8. Verdenelli, M.; Parola, S.; Hubert-Pfalzgraf, L. G.; Lecocq, S.; Polyhedron 2000, 19, 2069.
- 9. Arias, J. L.; Cabrera, A.; Sharma, P.; Rosas, N.; Garcia, J. L.; Hernandes, S.; Inorg. Chim. Acta 2000, 310, 261.
- 10. Buemi, G.; Zuccarello, F.; Chem. Phys. 2004, 306, 115.
- 11. Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V.; J. Am. Chem. Soc. 1989, 111, 1023.
- 12. Gilli, G.; Bertolasi, V.; Ferretti, V.; Gilli, G.; J. Am. Chem. Soc 1994, 116, 909.
- 13. Pimentel, G. C.; McClellan, A. L.; The Hydrogen Bond, 1st ed, W. H. Freeman and Company, Reinhold Publishing Corporation: New York, 1960.
- 14. Madsen, G. K. H.; Iversen, B. B.; Larsen, F. K.; Kapon, M.; Reisner, G. M.; Herbstein, F. H.; J. Am. Chem. Soc 1998, 120, 10040.
- 15. Gilli, G.; Gilli, P.; J. Mol. Struct. 2000, 552, 1.
- 16. Rios, M. A.; Rodriguez, J.; J. Mol. Struct. (Theochem) 1991, 228, 149.
- 17. Buemi, G.; Zuccarello, F.; J. Mol. Struct. (Theochem) 2002, 581, 71.
- 18. Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G.; J. Am. Chem. Soc. 1991, 113, 4917.
- 19. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E. Jr.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Radbuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, L.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T. A.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A.; Gaussian 98, Gaussian Inc.: Pittsburgh PA, 1998.
- 20. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 37, 785.
- 21. Foresman, J. B.; Frisch, E.; Exploring Chemistry with Electronic Structure Methods, 2nd ed., Gaussian, Inc.: Pittsburgh, 1996.
- 22. Cuma, M.; Scheiner, S.; Kar, T.; J. Mol. Struct (Theochem) 1999, 467, 37.
- 23. Grabowski, S. J.; J. Mol. Struct. 2001, 562, 137.
- 24. Munn, R.W.; Eckhardt, C. J.; J. Phys. Chem. A 2001, 105, 6938.
- 25. Grabowski, S. J.; J. Phys. Org. Chem. 2004, 17, 18.
- 26. Gilli, P.; Bertolasi, V.; Pretto, L.; Lycka, A.; Gilli, G.; J. Am. Chem. Soc. 2002, 124, 13554.
- 27. Pauling, L.; J. Am. Chem. Soc 1947, 69, 542;
- Pauling, L.; The Nature of the Chemical Bond, 3rd ed., Cornell University Press: Ithaca, NY, 1960.
- ; Bürgi, H.-B.; Dunitz, J. D.; J. Am. Chem. Soc 1987, 109, 2924.
- 28. Reed, A. E.; Weinstock, R. B.; Weinhold, F.; Chem. Phys. 1985, 83, 735.
- 29. Gilli, P.; Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, G.; J. Am. Chem. Soc. 2004, 126, 3845.
- 30. Trendafilova, N.; Bauer, G.; Mihaylov, T.; Chem. Phys. 2004, 302, 95.
Datas de Publicação
-
Publicação nesta coleção
06 Set 2011 -
Data do Fascículo
Dez 2006
Histórico
-
Aceito
21 Fev 2006 -
Recebido
02 Ago 2005