Resumo
alpha-Bromoacetophenones are important in organic synthesis. They have been widely used as precursors of several natural products. Several methods of bromination aiming at their synthesis have been described, however they furnish a mixture of starting material 10, mono 8 and dibromide 11 products. We developed a novel, simple and efficient synthesis of these compounds with applications in the synthesis of benzoylbenzofurans 9, compounds with important pharmacological properties, such as the ability of dilating the coronary artery and analgesic action. Such compounds have also been used as key intermediates to obtain quinone systems.
alpha-bromoacetophenones; benzoylbenzofurans; phenyltrimethylammonium tribromide
alpha-bromoacetophenones; benzoylbenzofurans; phenyltrimethylammonium tribromide
ARTIGO
Uma nova síntese de a-bromo-acetofenonas e sua aplicação na obtenção de 2-benzoil-benzofuranas
A novel synthesis of a-bromoacetophenones and its application in obtaining 2-benzoylbenzofuranes
Mariangela S. Azevedo; Ana Paula L. Alves; Glaucia B. C. Alves; Jari N. Cardoso; Rosângela S. C. Lopes; Cláudio C. Lopes* * e-mail: iqg02022@acd.ufrj.br
Departamento de Química Analítica, Instituto de Química, Universidade Federal do Rio de Janeiro, CT, Bloco A, 21949-900 Rio de Janeiro - RJ, Brasil
ABSTRACT
a-Bromoacetophenones are important in organic synthesis. They have been widely used as precursors of several natural products. Several methods of bromination aiming at their synthesis have been described, however they furnish a mixture of starting material 10, mono 8 and dibromide 11 products.
We developed a novel, simple and efficient synthesis of these compounds with applications in the synthesis of benzoylbenzofurans 9, compounds with important pharmacological properties, such as the ability of dilating the coronary artery and analgesic action. Such compounds have also been used as key intermediates to obtain quinone systems.
Keywords: a-bromoacetophenones; benzoylbenzofurans; phenyltrimethylammonium tribromide.
INTRODUÇÃO
Os sistemas benzofurânicos estão amplamente distribuídos na natureza. Sua origem biossintética foi proposta por alguns pesquisadores a partir do metabolismo secundário do ácido xiquímico1. Suas propriedades químicas e atividades biológicas têm despertado um interesse crescente de grupos de pesquisa, tanto no ramo da fitoquímica, quanto no da química orgânica sintética.
Desde o início da década de 60, as benzofuranas vêm sendo isoladas de muitos vegetais de grande porte, tais como de algumas espécies das famílias Rutaceae, Liliaceae, Cyperaceae e, principalmente, da Asteraceae2.
As amoreiras, plantas do gênero Morus, são largamente cultivadas na China, Japão e outros países. As cascas das raízes, bem como suas folhas, são tradicionalmente utilizadas na medicina popular asiática, como diurético, expectorante e laxante3. As atividades fungicida, bactericida e hipoglicemiante das folhas de Morus Alba e Morus insignis4 têm sido atribuídas às benzofuranas Mulberrofurana U e Moracin M-3'-O-b-D-glucopiranosídeo.
As 2- e 3-arilbenzofuranas 1 e 2 (Figura 1), sintetizadas por Kuo e Ross5, são agentes antiarrítmicos, antianginais, anti-hipertensivos, anti-inflamatórios e anti-hiperlipidêmicos6.
As benzofuranas, além de suas propriedades biológicas, constituem intermediários-chave na obtenção de sistemas policíclicos naturais. Dentre estes, encontram-se as benzoil-benzofuranas, compostos com significativas propriedades farmacológicas.
Buu-Hoïe colaboradores7, ao estudarem os efeitos causados pela 2-(4-hidroxi)-benzoil-3-etil-benzofurana e 2-etil-3-(3-hidroxi)-benzoil-benzofurana em cobaias, observaram que tais compostos exerciam efeito relaxante no espasmo causados pela histamina e a acetilcolina, bem como na mobilidade de cobaias. A amidarona (3), por sua vez, causa a dilatação de coronárias, apresentando, portanto, propriedades anti-anginais8 (Figura 1).
Devido ao grande interesse nos sistemas benzofurânicos, vários grupos de pesquisa têm dedicado esforços na síntese destes compostos. O interesse pelas benzofuranas vem de longo tempo, estando voltado principalmente, para as investigações referentes às suas atividades farmacológicas e à sua reatividade química.
Rap9 obteve previamente núcleos benzoilbenzofurânicos, porém seus rendimentos não foram citados.
Dai e Lai10, em uma reação "one-pot" via acoplamento-cruzado com iodonitrofenol em tolueno na presença de paládio, sintetizaram a benzoil-benzofurana (2) em rendimentos satisfatórios. No entanto, as condições utilizadas inviabilizavam sua síntese em larga escala.
Ono e colaboradores11 obtiveram o anel benzoilbenzofurânico através do acoplamento cruzado do 1-bromo-3,4-dimetoxibenzeno, nas condições de Negishi, tendo como catalisador PdCl2(dppf).
Roger e colaboradores12, ao investigarem as atividades farmacológicas destes compostos frente aos receptores estrogênicos, prepararam uma série de benzofuranas em fase sólida, através do tratamento de 2-hidroxi-benzaldeído com 7 equivalentes da a-bromo-acetofenona correspondente, na presença de DBU sob refluxo. Os rendimentos das aril-benzofuranas obtidas não foram mencionados.
Neste trabalho apresentamos uma nova rota sintética, simples e eficiente, de obtenção das a-bromo-acetofenonas 4a-h, com aplicação na síntese de 2-benzoil-benzofuranas 5a-s, viabilizando-a para futuros testes farmacológicos.
PARTE EXPERIMENTAL
Procedimentos experimentais gerais
Nas reações sensíveis à presença de umidade, utilizou-se vidraria seca em estufa a 140-150 ºC, por no mínimo 4 h, sendo posteriormente protegida do contato com a atmosfera por septos, e resfriada à temperatura ambiente sob pressão positiva de argônio.
Todos os solventes utilizados para fins sintéticos foram adquiridos com grau de pureza analítica. Os solventes utilizados nas reações foram previamente purificados e secos, de acordo com os procedimentos descritos na literatura13.
O bromo elementar, brometo de cobre II e tribrometo de feniltrimetilamônio foram obtidos comercialmente das empresas Merck Indústrias Química S. A., Aldrich e Fluka Chemica. O orto-hidroxibenzaldeído e o etilenoglicol foram obtidos comercialmente da Vetec.
Todas as reações foram monitoradas por cromatografia em camada delgada (CCD), empregando cromatofolhas de alumínio em sílica gel 60 F254(Merck), com espessura de 0,2 mm.
Para a purificação dos compostos, utilizou-se a cromatografia em coluna de sílica gel 60 (230-400 mesh ASTM Merck), com o sistema de solventes adequado em cada caso, caracterizando uma cromatografia do tipo "flash"14.
Os espectros na região do infravermelho, obtidos em espectrômetro Magna IR-760-Nicolet, foram registrados utilizando-se pastilhas de KBr para os compostos sólidos, ou como filme líquido no caso de amostras oleosas. Os valores para as absorções foram referidos em número de ondas, utilizando como unidade o centímetro recíproco (cm-1).
Espectros de ressonância magnética nuclear de hidrogênio (RMN1H) e de carbono (RMN13C), com desacoplamento completo de prótons, foram obtidos em espectrômetro Varian modelo Gemini-200 MHz-FT ou Bruker-300 MHz, utilizando o tetrametilsilano (TMS) como referência interna, e CDCl3 como solvente. Os deslocamentos químicos (d) foram referidos em parte por milhão (ppm), em relação ao TMS, e as constantes de acoplamento (J), em Hertz (Hz). Os dados espectrométricos são reportados da seguinte forma: s = sinal simples; d = sinal duplo; = sinal triplo, m = sinal múltiplo; l = sinal largo; 2s = dois sinais simples; dd = duplo sinal duplo; ddd = duplo duplo sinal duplo; dt= duplo sinal triplo; dq =duplo quádruplo).
Os espectros de massa (EM) foram obtidos em cromatógrafo gasoso acoplado a espectrômetro de massas, usando o aparelho do tipo HP modelo 6890, acoplado a detector seletivo de massas modelo G530A.
Os pontos de fusão foram determinados em aparelho MELT-TEMPII.
Procedimentos sintéticos
Síntese de a-bromo-acetofenonas (4a-f)
Procedimento A: Br2/éter
Em um balão tritubulado, adicionou-se a metil fenil cetona 6a (168,0 mg; 1,40 mmoles) em 10 mL de éter etílico anidro. A solução foi mantida sob agitação e resfriada a 0 ºC. Em seguida, adicionou-se 5 mL de uma solução contendo Br2 (1,33 mg; 8,30 mmoles) em 5 mL de éter etílico anidro. A mistura reacional foi mantida sob agitação, a 0 ºC, sob atmosfera de argônio por 5 h, sendo a reação acompanhada por CCD. Ao término da reação, adicionou-se 10 mL de solução aquosa de bissulfito de sódio a 5%, para eliminar o excesso de bromo. O produto foi extraído com 3 x 10 mL de éter etílico e a fase orgânica lavada com água (3 x 10 mL) até pH neutro, foi seca com Na2SO4. O produto bruto foi obtido após evaporação do solvente, e a purificação foi feita por cromatografia em sílica, utilizando como eluente EtOAc/Hexano 1, 2 e 5% (100 mL de cada).
Procedimento B: CuBr2/CHCl3/EtOAc
Em um balão tritubulado equipado com agitador magnético e condensador de refluxo, sob atmosfera de argônio, adicionou-se CuBr2 (447,00 mg; 2,00 mmoles), em 20 mL de clorofórmio anidro. Após refluxo brando, acrescentou-se uma solução contendo a acetofenona 6a (120,00 mg; 1,00 mmol), em 10 mL acetato de etila anidro. A mistura reacional foi refluxada até que a cor verde resultante passasse a âmbar, e cessasse a evolução de HBr. A reação foi resfriada, filtrada, e o resíduo inorgânico foi lavado com 3 x 20 mL de acetato de etila. A mistura de solventes orgânicos foi lavada com água destilada (3 x 25 mL) até pH neutro. A fase orgânica foi seca sobre Na2SO4 e evaporada em rota evaporador. O produto bruto foi purificado em coluna, utilizando como eluente EtOAc/Hexano 1, 2 e 5%.
Procedimento C: Etilenoglicol/ C6H5N+(CH3 )3Br3-/THF
Em um balão de fundo redondo com agitador magnético e septo, sob atmosfera de argônio, dissolveu-s acetofenona 6a (288,00 mg; 2,40 mmoles) em 3 mL de THF anidro e 5 mL de etilenoglicol. A esta solução, adicionou-se tribrometo de feniltrimetilamônio (1,84 g; 4,90 mmoles) em 30 mL de THF anidro. A mistura reacional foi agitada por 20 h, à temperatura ambiente, e acompanhada por CCD. O meio reacional foi neutralizado através da adição de uma solução gelada de NaHCO3 10%, seguida de 6 mL de tiossulfato de sódio 5% (m/v) e, finalmente, a fase orgânica foi extraída com 2 x 40 mL de éter etílico. O extrato etéreo foi lavado com uma solução saturada de NaCl (3 x 15 mL), até pH neutro. A fase orgânica foi seca sobre Na2SO4 e os solventes evaporados. O produto bruto foi purificado em coluna, utilizando como eluente EtOAc/Hexano 1, 2 e 5%, com exceção de 8b (eluente - CHCl3).
a-bromo-acetofenona (4a): Procedimento A: (139,30 mg; 0,70 mmoles) 50% de (4a) (116,76 mg; 0,42 mmoles) 30% de (7a); Procedimento B: (179,10 mg; 0,90 mmoles) 90% de (4a) e (13,90 mg; 0,05 mmoles) 5% de (7a); Procedimento C: (334,32 mg; 1,68 mmoles) 70% de (4a) e (33,36 mg; 0,12 mmoles) 5% de (7a). Dados de 4a: p.f. 47 ºC (lit. 48-51 °C)15. IV (KBr, cm-1)  : 1674 (CO). RMN1H (200 MHz, CDCl3) d: 4,46 (s, 2H, CO-CH2-Br); 7,52- 7,58 (m, 3H); 7,92- 7.98 (m, 2H).
: 1674 (CO). RMN1H (200 MHz, CDCl3) d: 4,46 (s, 2H, CO-CH2-Br); 7,52- 7,58 (m, 3H); 7,92- 7.98 (m, 2H).
a-orto-dibromo-acetofenona (4b) e a,a-,orto-tribromo-acetofenona (7b): Procedimento B: (222,40 mg; 0,80 mmoles) 80% de (4b) e (17,85 mg; 0,05 mmoles) 5% de (7b); Procedimento C: (507,07 mg; 1,82 mmoles) 76% de (4b) e (59,98 mg; 0,17 mmoles) 7% de (7b). Dados de 4b: óleo incolor. IV (filme, cm-1)  max: 1673 (CO). RMN1H (200 MHz, CDCl3) d: 4,49 (s, 2H, CO-CH2-Br); 7,20 (m, 3H); 7,63 (q, 1H). RMN13C (50 MHz, CDCl3) d: 191,59 (CO); 138,66; 132,71; 130,06; 129,68; 127,50; 119,06; 33,70 (CH2). EM, m/z (%): 287 (M+, 4), 185 (100), 184 (98), 156 (20), 155 (20). Dados de 7b: óleo incolor. RMN1H (200 MHz, CDCl3) d: 6,71 (s, 1H, CO-CH-Br); 7,25-7,45 (m, 2H); 7,51-7,65 (m, 2H).
max: 1673 (CO). RMN1H (200 MHz, CDCl3) d: 4,49 (s, 2H, CO-CH2-Br); 7,20 (m, 3H); 7,63 (q, 1H). RMN13C (50 MHz, CDCl3) d: 191,59 (CO); 138,66; 132,71; 130,06; 129,68; 127,50; 119,06; 33,70 (CH2). EM, m/z (%): 287 (M+, 4), 185 (100), 184 (98), 156 (20), 155 (20). Dados de 7b: óleo incolor. RMN1H (200 MHz, CDCl3) d: 6,71 (s, 1H, CO-CH-Br); 7,25-7,45 (m, 2H); 7,51-7,65 (m, 2H).
a-bromo-meta-metoxi-acetofenona (4c): Procedimento C: (494,64 mg; 2,16 mmoles) 90% de (4c). Dados de 4c: sólido amarelo, p.f. 71,5 ºC (etanol/hexano), lit. 72.0 ºC15. IV (KBr, cm-1) max:1634 (CO). RMN1H (300 MHz, CDCl3) d: 3,78 (s, 3H, OCH3); 4,33 (s, 2H, CO-CH2-Br); 7,06 (ddd, J = 1,0 Hz, J = 2,6 Hz, J = 8,2 Hz, 1H); 7,32 (t, J = 7,7 Hz, 1H); 7,43(d, J = 2,5 Hz, 1H); 7,49 (dd, J = 1,0 Hz, J = 9,0 Hz, 1H). RMN13C (75 MHz, CDCl3) d: 160,14; 135,93; 129,99; 121,64; 120,66; 113,26; 55,67 (OCH3); 31,19 (CO-CH2-Br).
a-bromo-para-metoxi-acetofenona (4d) e a,a-dibromo-para-metoxi-acetofenona (7d): Procedimento A: a partir de (I,030 mg; 6,90 mmoles) 10d e (1,100 mg; 6,90 mmoles) de Br2. (361,84 mg; 1,58 mmoles) 22,9% de (4d) e (637,56 mg; 2,07 mmoles) 30% de (7d); Procedimento B:(178,62 mg; 0,78 mmoles) 78% de (4d) e (30,80 mg; 0,10 mmoles) 10% de (7d); Procedimento C: (467,16 mg; 2,04 mmoles) 85% de (4d) e (22,18 mg; 0,07 mmoles) 3% de (7d). Dados de 4d: sólido amarelo, p.f. 65,5-66,5 ºC (CH2Cl2)15. IV (KBr, cm-1) max:1650 (CO). RMN1H (300 MHz, CDCl3,) d: 3,90 (s, 3H, OCH3); 4,40 (s, 2H, CO-CH2-Br); 6,98 (d, J = 10,0 Hz, 2H); 7,98 (d, J = 10,0 Hz, 2H). EM, m/z (%): 229 (M+, 7), 228 (8), 135 (100), 107 (7), 92 (12). Dados de 7d: p.f. 90,5-92,5 ºC (hexano/acetato). IV (KBr, cm-1) max: 1679 (CO). RMN1H (300 MHz, CDCl3) d: 3,90 (s, 3H, OCH3); 6,68 (s, 1H, CO-CH-Br); 6,98 (d, J = 10,0 Hz, 2H); 8,09 (d, J = 10,0 Hz, 2H). EM, m/z (%): 308 (M+, 35), 201 (40), 200 (4), 199 (42), 186 (70); 156 (12).
a-bromo-3,4-dimetoxi-acetofenona (4e) e a,a-dibromo-3,4-dimetoxi-acetofenona (7e): Procedimento C: (435,12 mg; 1,68 mmoles) 70% de (4e) e (56,78 mg; 0,17 mmoles) 7% de (7e). Dados de 4e: sólido laranja, p.f. 75,5 ºC (hexano)32. IV (KBr, cm-1) max:1662 (CO). RMN1H (300 MHz, CDCl3) d: 3,95 (s, 3H, OCH3); 3,96 (s, 3H, OCH3); 4,41 (s, 2H, CO-CH2-Br); 6,91 (d, J = 8,4 Hz, 1H,); 7,55 (d, J = 1,8 Hz, 1H); 7,62 (dd, J = 1,9 Hz, J = 6,5 Hz, 1H). RMN13C (75 MHz, CDCl3) d: 190,25 (CO); 154,18; 149,46; 127,20; 124,20; 110,98; 110,25; 56,32 (OCH3); 56,20 (OCH3); 30,22 (CO-CH2-Br). EM, m/z (%): 260 (M+, 11), 259 (11), 165 (100). Dados de 7e: RMN1H (300 MHz, CDCl3) d: 3,84 (s, 3H, OCH3); 3,86 (s, 3H, OCH3); 6,62 (s, 1H, CO-CH-Br); 6,91 (d, J = 8,4 Hz, 1H); 7,55 (d, J = 1,8 Hz, 1H); 7,62 (dd, J = 1,9 Hz, J = 6,5 Hz, 1H). RMN13C (75 MHz, CDCl3) d: 184,98; 154,66; 149,56; 124,55; 123,63; 56,38 (OCH3); 56,25 (OCH3); 39,72 (CH-Br).
a-bromo-3,4-metilenodioxi-acetofenona (4f) e a,a-dibromo-3,4-metilenodioxi-acetofenona (7f): Procedimento A: a partir de (500,20 mg; 3,05 mmoles) 6f e de (488,00 mg; 3,05 mmoles) Br2 (503,98 mg; 2,07 mmoles) 68% de (4f) e (78,57 mg; 0,24 mmoles) 8% de (7f). Procedimento B: (191,97 mg; 0,79 mmoles) 79% de (4f) e (41,86 mg; 0,13 mmoles) 13% de (7f); Procedimento C: (524,88 mg; 2,16 mmoles) 90% de (4f) . Dados de 4f: sólido amarelo, p.f. 90,5-91,0 ºC (acetato/hexano). IV (KBr, cm-1) max: 1684 (CO). RMN1H (200 MHz, CDCl3) d: 4,37 (s, 2H, CO-CH2-Br), 6,07 (s, 2H, O-CH2-O), 6,87 (d, J = 8,0 Hz, 1H), 7,45 (d, J = 1,7 Hz, 1H), 7,58 (dd, J = 1,7, J = 8,0 Hz, 1H). RMN13C (75 MHz, CDCl3) d: 180,00 (CO); 112,45; 111,71; 109,70; 105,34; 101,01; 99,07; 95,07, 26,50 (OCH2). EM, m/z (%): 243 (M+, 11), 242 (11), 149 (100), 121 (17). Dados de 11f: p.f.: 80-83 ºC (CH2Cl2). EM, m/z (%): 324(0,5), 322 (1), 320(M+, 0,5), 149(100).
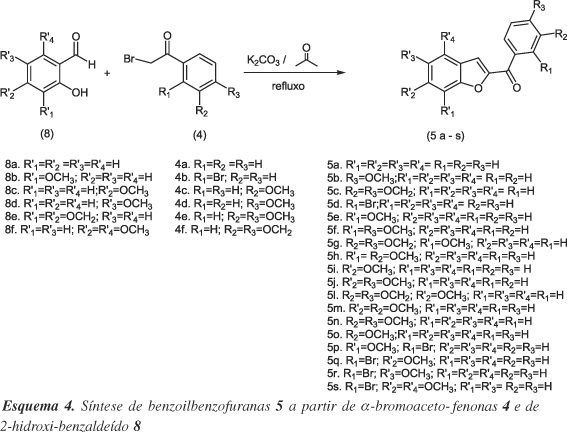
Síntese de 2-benzoil-benzofuranas (5a-s)
Em um balão tritubulado, provido de um condensador de refluxo, contendo 2-hidroxibenzaldeído (8a) (317,20 mg; 2,60 mmoles) em 10 mL de acetona anidra, e K2CO3 (358,80 mg; 2,60 mmoles), mantidos sob refluxo por 15 min, adicionou-se, gota a gota, uma solução de a-bromo-acetofenona (4a) (497,50 mg; 2,50 mmoles) em 10 mL de acetona anidra. A mistura resultante foi refluxada por 4 h. A reação foi realizada sob atmosfera de argônio e monitorada por CCD. Ao término da reação, resfriou-se o balão e a mistura reacional foi filtrada; o produto da reação foi extraído do resíduo após adição de 3 x 20 mL de acetato de etila. A fase orgânica coletada foi lavada com uma solução saturada de NaCl (3 x 15 mL) até pH neutro, seca com Na2SO4 e evaporada em rota evaporador. O material bruto foi purificado em coluna, utilizando como eluente EtOAc/hexano 10%.
2-benzoil-benzofurana (5a): (388,50 mg; 1,75 mmol) 70% de (5a). sólido amarelo, p.f. 89,5-90 ºC (CH2Cl2/hexano). IV (KBr, cm-1) : 1640 (CO). RMN1H (200 MHz, CDCl3) d: 7,30-7,32 (m, 1H); 7,47-7,76 (m, 7H); 8,03 (d, J = 1,3 Hz, 2H); 8,06 (d, J = 1,6 Hz, 1H). EM, m/z (%): 222 (M+, 55), 105 (60), 145 (27), 77 (100). Análise elementar calculada (%): C (81,08), H (4,50) ; experimental (%): C (81,66), H (4,50).
2-(4-metoxi)-benzoil-benzofurana (5b): a partir de (122,00 mg; 1,00 mmol) 2-hidroxi-benzaldeído (8a) e de (229,00 mg; 1,00 mmol) a-bromo-para-metoxi-acetofenona (4d). (178,92 mg; 0,71 mmoles) 71% de (5b). sólido amarelo, p.f. 90-91 ºC (acetato de etila/hexano). IV (KBr, cm-1) max: 1650 (CO). RMN1H (200 MHz, CDCl3) d: 3,89 (s, 3H, OCH3); 6,98 (d, J = 11,98 Hz, 1H); 6,99 (d, J = 5,0 Hz, 1H); 7,27 (m, 1H); 7,42 (d, J = 0,7, 1H); 7,44 (s, 1H); 7,57 (d, J = 0,8 Hz, 1H); 7,61 (d, J = 0,6 Hz, 1H); 8,07 (d, J = 5,0 Hz, 1H); 8,11 (d, J = 11,98 Hz, 1H). RMN13C (50 MHz, CDCl3,) d: 163,51; 162,44; 155,66; 152,71; 131,76; 129,73; 127,77; 125,33; 124,15; 122,97; 115,11; 113,72; 112,24; 55,32 (OCH3). EM, m/z (%): 252 (M+, 53), 237 (8), 221 (22), 135 (100), 107 (14); Análise elementar calculada (%): C (76,19), H (4,76); experimental (%): C (76,70), H (4,83).
2-(3,4-metilenodioxi)-benzoil-benzofurana (5c): a partir de (122,00 mg; 1,00 mmol) 2-hidroxi-benzaldeído (8a) e de (243,00 mg; 1,00 mmol) a-bromo-3,4-metilenodioxi-acetofenona (4f). (239,40 mg: 0,90 mmoles) 90% de (5c). sólido vermelho, p.f. 80-81 ºC (acetato de etila/hexano). IV (KBr, cm-1) max: 1660 (CO). RMN1H (200 MHz, CDCl3) d: 6,01 (s, 2H, OCH2O); 6,85 (d, J = 8,15 Hz, 1H); 7,27 (t, J = 7,6 Hz, 1H); 7,43 (t, J = 8,4 Hz, 1H); 7,46 (s, 1H); 7,51 (t, J = 8,4 Hz, 1H); 7,56 (d, J = 1,5 Hz, 1H); 7,65 (d, J = 7,3 Hz, 1H); 7,71 (dd, J = 1,7 Hz, J = 6,9 Hz, 1H). RMN13C (50 MHz, CDCl3) d: 182,13 (CO); 155,74; 152,38; 151,87; 148,02; 131,32; 128,06; 126,00; 123,88; 123,16; 115,63; 112,21; 109,38; 107,96; 101,96; 26,8 (OCH2O). EM, m/z (%): 266 (M+, 69), 149 (100), 119 (28), 237 (15). Análise elementar calculada (%): C (72,18), H (3,76); experimental C (72,57), H (4,02).
2-(2-bromo)-benzoil-benzofurana (5d): a partir de (122,00 mg; 1,00 mmol) 2-hidroxi-benzaldeído (8a) e de (278,00 mg; 1,00 mmol) a-orto-dibromo-acetofenona (4b). (255,85 mg; 0,85 mmoles) 85% de (5d). Óleo amarelo. IV (filme, cm-1) max: 1640 (CO). RMN1H (200 MHz, CDCl3) d: 7,47 (s, 1H); 7,26-7,70 (m, 11H). RMN13C (50 MHz, CDCl3) d: 184,76 (CO); 156,57; 151,76; 139,50; 133,65; 132,00; 129,39; 129,16; 127,32; 127,13; 124,29; 123,73; 120,12; 118,17; 112,85. EM, m/z (%): 302 (M+, 74), 301 (74), 221 (33), 183 (46), 145 (100). Análise elementar (%) calculada C (59,80), H (2,99), experimental C (60,12), H (2,97).
2-benzoil-7-metoxi-benzofurana (5e): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-3-metoxi-benzaldeído (8b) e de (199,00 mg; 1,00 mmol) a-bromo-acetofenona (4a). (191,52 mg; 0,76 mmoles) 76% de (5e). sólido laranja, p.f. 58-59 ºC (CH2Cl2). IV (KBr, cm-1) max: 1641 (CO). RMN1H (200 MHz, CDCl3) d: 4,03 (s, 3H, OCH3); 6,96 (dd, J = 1,6 Hz, J = 7,3 Hz, 1H); 7,26 (m, 2H); 7,50 (s,1H); 7,58 (m, 3H); 8,06 (d, J = 1,7 Hz, 1H); 8,11 (d, J= 1,2 Hz, 1H). RMN13C (50 MHz, CDCl3) d: 183,80 (CO); 152,93; 146,33; 145,97; 137,35; 132,83; 129,62; 128,88; 128,53; 124,68; 116,02; 115,12; 110,20; 56,39. EM, m/z (%): 252 (M+, 100), 175 (27), 105 (84). Análise elementar calculada (%): C (76,19), H (4,76); experimental (%): C (76,57), H (4,85).
2-(4-metoxi)-benzoil-7-metoxi-benzofurana (5f): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-3-metoxi-benzaldeído (8b) e de (229,00 mg; 1,00 mmol) a-bromo-para-metoxi-acetofenona (4d). (253,80 mg; 0,90 mmoles) 90% de (5f). sólido laranja, p.f. 78-79,5 ºC (hexano). IV (KBr, cm-1) max: 1650 (CO). RMN1H (200 MHz, CDCl3) d: 3,88 (s, 3H, OCH3); 4,03 (s, 3H, OCH3); 6,95 (dd, J = 1,5 Hz, J = 6,0 Hz, 1H); 6,99 (dd, J = 3,0 Hz, J = 9,0 Hz, 1H); 7,01 (dd, J = 2,0 Hz, J = 5,0 Hz, 1H); 7,22 (dd, J = 7,9 Hz, J = 9,5 Hz, 1H); 7,24 (dd, J = 1,5 Hz, J = 6,0 Hz, 1H); 7,50 (s, 1H); 8,13 (dd, J = 1,8 Hz, J = 6,8 Hz, 1H); 8,15 (dd, J = 2,8 Hz, J = 11,6 Hz, 1H). RMN13C (50 MHz, CDCl3) d: 182,15 (CO); 163,56; 153,17; 146,03; 145,48; 131,98; 129,71; 128,66;. 124,42; 115,07; 114,80; 113,77; 109,46; 56,07 (OCH3); 55,37 (OCH3). EM, m/z(%): 282 (M+, 72), 175 (3), 135 (100). Análise elementar calculada (%): C (72,34), H (4,96); experimental (%): C (72,69), H (5,01).
2-(3,4-metilenodioxi)-benzoil-7-metoxi-benzofurana (5g): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-3-metoxi-benzaldeído (8b) e de (243,00 mg; 1,00 mmol) a-bromo-3,4-metilenodioxi-acetofenona (4f). (207,20 mg; 0,70 mmoles) 70% de (5g). sólido amarelo, p.f. 95-95,5 ºC (hexano). IV (KBr, cm-1) max: 1631 (CO). RMN1H (200 MHz, CDCl3) d: 4,03 (s, 3H, OCH3); 6,90 (d, J = 7,9 Hz, 1H); 6,94 (d, J = 1,3 Hz, 1H); 7,22 (t, J = 7,6 Hz, 1H); 7,24 (dd, J = 0,9 Hz, J = 7,4 Hz, 1H); 7,49 (s, 1H); 7,60 (d, J = 1,6 Hz, 1H); 7,78 (dd, J = 1,7 Hz, J = 7,0 Hz, 1H). RMN13C (50 MHz, CDCl3) d: 181,79; 153,27; 152,00; 148,22; 146,33; 131,68; 128,94; 126,32; 124,62; 115,28; 115,04; 110,04; 109,75; 108,13; 101,92; 56,41 (OCH3); 29,76 (OCH2O). EM, m/z (%): 296 (M+, 75), 175 (24), 149 (100), 121 (25). Análise elementar calculada (%): C (68,92), H (4,05); experimental (%): C (69,02), H (4,24).
2-(3-metoxi)-benzoil-7-metoxi-benzofurana (4h): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-3-metoxi-benzaldeído (8b) e de (229,00 mg; 1,00 mmol) a-bromo-meta-metoxi-acetofenona (4c). (250,98 mg; 0,89 mmoles) 89% de (5h). Óleo incolor. IV (filme, cm-1) : 1649 (CO). RMN1H (200 MHz, CDCl3) d: 3,87 (s, 3H, OCH3); 4,02 (s, 3H, OCH3); 6,95 (dd, J = 1,5 Hz, J = 7,1 Hz, 1H); 7,11-7,20 (m, 1H); 7,23-7,50 (m, 1H); 7,40 (t, J = 7,0 Hz, 1H); 7,52 (s, 1H); 7,58- 7,62 (m, 1H); 7,68- 7,72 (m, 1H). RMN13C (50 MHz, CDCl3) d: 184,20 (CO); 176,24; 164,24; 152,32; 129,98; 127,85; 125;74; 119,54; 117,24; 115,43; 112,57; 111,34; 110,00; 108,72; 107,54; 55,30 (OCH3); 54,20 (OCH3). EM, m/z (%): 282 (M+, 100), 251 (12), 175 (34), 135 (63), 107 (37). Análise elementar calculada (%): C (72,34), H (4,96); experimental (%): C (72,54), H (5,06).
2-benzoil-6-metoxi-benzofurana (5i): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-4-metoxi-benzaldeído (8c) e de (199,00 mg; 1,00 mmol) a-bromo-acetofenona (4a). (214,20 mg; 0,85 mmoles) 85% de (5i). sólido branco, p.f. 93-95 ºC (éter/CH2Cl2). IV (KBr, cm-1) max: 1665 (CO). RMN1H (200 MHz, CDCl3) d: 3,86 (s, 3H, OCH3); 6,92 (dd, J = 2,2 Hz, J = 8,6 Hz, 1H); 7,07 (d, J = 2,2 Hz, 1H); 7,42-7,62 (m, 5H); 7,99 (d, J = 1,6 Hz, 1H); 8,02 (t, J = 1,7 Hz, 1H). RMN 13C (50 MHz, CDCl3) d: 193,43 (CO); 183,70; 161,43; 157,77; 152,28; 137,78; 132,56; 129,38; 128,50; 123,63; 120,60; 114,54; 95,98; 55,82 (OCH3). EM, m/z (%) 252 (M+, 100), 237 (12), 175 (34), 105 (67). Análise elementar calculada (%): C (76,19), H (4,76); experimental (%): C (76,64), H (4,79).
2-(4-metoxi)-benzoil-6-metoxi-benzofurana (5j): a partir de (998,60 mg; 6,57 mmoles) 2-hidroxi-4-metoxi-benzaldeído (8c), 5 mL de acetona anidra e de (1,505 mg; 6,57 mmoles) a-bromo-para-metoxi-acetofenona (4d). O material bruto foi purificado em coluna, utilizando EtOAc/ hexano 10%. (1,570 mg; 5,58 mmoles) 85% de (5j). sólido laranja, p.f. 140-142 ºC (hexano). IV(KBr, cm-1)
max:1600 (CO). RMN1H (200 MHz, CDCl3) d: 3,89 (s, 3H, OCH3); 3,91 (s, 3H, OCH3); 6,98 (d, J = 8,7 Hz, 1H); 7,04 (d, J = 9,0 Hz, 2H); 7,14 (d, J = 1,9 Hz, 1H); 7,48 (d, J = 0,6 Hz, 1H); 7,60 (d, J = 8,7 Hz, 1H), 8,05 (d, J = 9,0 Hz, 2H). RMN 13C (50 MHz, CDCl3) d: 182,17 (CO); 163,25; 160,84; 157,26; 152,22; 131,58; 130,01; 129,01; 123,27; 120,30; 115,91; 114,13; 113,64; 95,58; 55,57 (OCH3); 55,33 (OCH3). EM, m/z (%): 282 (M+, 100), 251 (7), 135 (49). Análise elementar calculada (%) C (72,34), H (4,96); experimental (%): C (72,72), H (5,15).2-(3,4-metilenodioxi)-benzoil-6-metoxi-benzofurana (5l): a partir de (998,64 mg; 6,57 mmoles) 2-hidroxi-4-metoxi-benzaldeído (8c), 5 mL de acetona anidra e de (1600 mg: 6,57 mmoles) a-bromo-3,4-metilenodioxi-acetofenona (4f). O material bruto foi purificado em coluna, utilizando EtOAc/ hexano 10%. (1540 mg; 5,26 mmoles) 80% de (5l). sólido vermelho, p.f. 144-145 ºC (hexano). IV (KBr, cm-1) max: 1600 (CO). RMN1H (50 MHz, CDCl3) d: 3,88 (s, 3H, OCH3); 6,08 (s, 2H, O-CH2-O); 6,90 (s, 1H); 6,98 (d, J = 8,0 Hz, 1H); 7,09 (s, 1H); 7,46 (d, J = 0,9 Hz, 1H); 7,53 (t, 1H); 7,59 (s, 1H); 7,69 (dd, J = 1,6 Hz, J = 6,4 Hz, 1H). EM, m/z (%): 296 (M+, 100), 266 (10), 149 (100), 121 (30). RMN 13C (200 MHz, CDCl3) d: 181,90 (CO); 161,02; 157,43; 151,97; 151,63; 147,97; 131,69; 125,67; 123,49; 120,31; 116,45, 114,39; 109,24; 107,99; 101,89; 96,23; 55,72 (OCH3); 30,33 (OCH3). Análise elementar calculada C (%) (68,92), H (4,05); experimental (%): C (68,99), H (4,12).
2-(3-metoxi)-6-metoxi-benzoil-benzofurana (5m): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-4-metoxi-benzaldeído (8c) e de (229,00 mg; 1,00 mmol) a-bromo-meta-metoxi-acetofenona (4c). (234,06 mg, 0,83 mmoles) 83% de (5m). sólido vermelho, p.f. 74-75 ºC (CH2Cl2). IV (KBr, cm-1) max: 1655 (CO). RMN1H (200 MHz, CDCl3) d: 3,87 (s, 3H, OCH3); 3,97 (s, 3H, OCH3); 6,96 (dd, J = 2,2 Hz; J = 8,7 Hz, 1H); 7,09 (ddd, J = 0,9 Hz; J = 1,7 Hz, J = 8,3 Hz, 1H); 7,41 (d, J = 1,9 Hz, 1H); 7,44 (t, J = 8,0 Hz, 1H); 7,46 (d, J = 0,9 Hz, 1H); 7,50 (m, 1H); 7,54 (s, 1H); 7,57 (t, J = 8,5 Hz, 1H). RMN13C (50 MHz, CDCl3) d: 161,29 (CO); 159,67; 157,78; 151,78; 138,81; 129,48; 123,67; 121,84; 120,36; 118,93; 117,41; 114,55; 113,89; 95,63; 55,74 (OCH3); 55,50 (OCH3). EM, m/z (%): 282 (M+, 100), 267 (9), 251 (13), 175 (39). Análise elementar calculada C (%) (72,34), H (4,96); experimental (%): C (72,46), H (5,27).
2-(3,4-dimetoxi)-benzoil-benzofurana (5n): a partir de (122,00 mg; 1,00 mmol) 2-hidroxi-benzaldeído (8a) e de (259,00 mg; 1,00 mmol) a-bromo-3,4-dimetoxi-acetofenona (4e). (239,70 mg; 0,85 mmoles) 85% de (5n). sólido amarelo-alaranjado, p.f. 60-62 ºC (hexano/acetato). IV (KBr, cm-1) max: 1659 (CO). RMN1H (300 MHz, CDCl3) d: 3,97 (s, 3H, OCH3); 3,98 (s, 3H, , OCH3); 6,95 (d, J = 8,4 Hz, 1H); 7,33 (d, J = 7,8 Hz, 1H); 7,35 (dt, J = 7,1 Hz, J = 0,8 Hz, 1H); 7,48 (dt, J = 6,0 Hz, J = 1,2 Hz, 1H); 7,55 (d, J = 0,84 Hz, 1H); 7,62 (d, J = 2,0 Hz, 1H); 7,65 (d, J = 0,7 Hz, 1H); 7,84 (dd, J = 2,0 Hz, J = 8,4 Hz, 1H). RMN13C (75 MHz, CDCl3) d: 182,90 (CO); 155,96; 153,59; 152,76; 149,28; 130,05; 128,18; 127,17; 124,67; 124,05; 123,30; 115,72; 112,60; 111,96; 110,97; 110,72; 110,23; 56,28 (OCH3); 56,24 (OCH3). EM, m/z (%): 282 (M+, 100), 251 (27), 165 (78), 145 (24).
2-(3-metoxi)-benzoil-benzofurana (5o): a partir de (122,00 mg; 1,00 mmol) 2-hidroxi-benzaldeído (8a) e de (229,00 mg; 1,00 mmol) a-bromo-meta-metoxi-acetofenona (4c). (176,40 mg; 0,70 mmoles) 70% de (5o). sólido laranja, p.f. 75-75,5 ºC (hexano). IV (KBr, cm-1) max: 1654 (CO). RMN1H (200 MHz, CDCl3) d: 3,88 (s, 3H, OCH3); 7,18 (ddd, J = 0,8 Hz, J = 2,5 Hz, J = 8,1 Hz, 1H); 7,30 (dt, J = 0,8 Hz, J = 7,1 Hz, 1H); 7,52 (m, 3H); 7,54 (s, 1H); 7,62 (s, 1H); 7,64 (d, J = 2,0 Hz, 1H); 7,73 (d, J = 7,7 Hz, 1H). RMN13C (50 MHz, CDCl3) d: 183,94 (CO); 159,97; 156,18; 152,60; 138,70; 129,57; 128,34; 127,17; 124,03; 123,34; 122,15; 119,39; 116,24; 114,32; 112,60; 55,59 (OCH3). EM, m/z (%): 252 (M+, 100), 237 (12), 221 (44), 135 (82), 107 (44). Análise elementar calculada C (%) (76,19), H (4,76); experimental (%):C (76,57), H (4,81).
2-(2-bromo)-benzoil-7-metoxi-benzofurana (5p): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-3-metoxi-benzaldeído (8b) e de (278,00 mg; 1,00 mmol) a-orto-dibromo-acetofenona (4b). (314,45 mg; 0,95 mmoles) 95% de (5p). Óleo incolor. IV ( filme, cm-1) max: 1660 (CO). RMN1H (200MHz, CDCl3) d: 3,80 (s, 3H, OCH3); 7,50 (d, J = 10,0 Hz, 1H); 7,707,40 (m, 7H). RMN13C (50 MHz, CDCl3) d: 184,80 (CO); 145,10; 139,20; 133,60; 131,90; 129,50; 127,30; 124,90; 121,60; 118,00; 115,30; 110,40; 56,20 (OCH3). EMAR m/z (M+) calculada para C16H11O3Br: 329,9892, encontrado: 329,9894.
2-(2-bromo)-benzoil-6-metoxi-benzofurana (5q): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-4-metoxi-benzaldeído (8c) e de (278,00 mg; 1,00 mmol) a-orto-dibromo-acetofenona (4b). (317,76 mg; 0,96 mmoles) 96% de (5q). sólido amarelo, p.f. 82 ºC. IV (KBr, cm-1)max: 1665 (CO). RMN1H (200 MHz, CDCl3) d: 3,90 (s, 3H, OCH3); 6,90 (dd, J = 6,0 e 3,0 Hz, 1H); 7,10 (s, 1H); 7,20 (s, 1H); 7,707,40 (m, 5H). RMN13C (50 MHz, CDCl3) d: 183,90 (CO); 161,80; 158,30; 151,40; 139,70; 133,60; 131,80; 129,30; 127,20; 124,00; 120,50; 120,10; 118,90; 114,90; 95,70; 55,90 (OCH3). EMAR m/z (M+) calculado para C16H11O3Br: 329.9892; encontrado 329,9889.
2-(2-bromo)-benzoil-5-metoxi-benzofurana (5r): a partir de (152,00 mg; 1,00 mmol) 2-hidroxi-5-metoxi-benzaldeído (8d) e de (278,00 mg; 1,00 mmol) a-orto-dibromo-acetofenona (4b). (314,45 mg; 0,95 mmoles) 95% de (5r). sólido amarelo, p.f. 76 ºC. IV (KBr, cm-1) max: 1660 (CO). RMN1H (300 MHz, CDCl3) d: 3,80 (s, 3H, OCH3); 7,10 (m, 2H); 7,507,40 (m, 5H); 7,70 (dd, J = 2,7 Hz e 12,0 Hz, 1H);. RMN13C (75 MHz CDCl3) d: 184,80 (CO); 157,10; 152,60; 152,00; 139,70; 133,90; 132,20; 129,70; 127,90; 127,60; 120,30; 119,70; 118,40; 113,70; 104,40; 56,20 (OCH3). EMAR m/z (M+) calculado para C16H11O3Br: 329,9892; encontrado 329,9861.
2-(2-bromo)-benzoil-4,6-dimetoxi-benzofurana (5s): a partir de (182,00 mg; 1,00 mmol) 2-hidroxi-4,6-dimetoxi-benzaldeído (8f) e de (278,00 mg; 1,00 mmol) a-orto-dibromo-acetofenona (4b). (353,78 mg; 0,98 mmoles) 98% de (5s). sólido amarelo, p.f. 99-100 ºC. IV (KBr, cm-1) max:1652 (CO). RMN1H (CDCl3, 200MHz) d: 3,90 (s, 6H, OCH3); 6,30 (s, 1H); 6,70 (s, 1H); 7,20 (s,1H); 7,507,40 (m, 3H); 7,70 (s, 1H). RMN13C (50 MHz, CDCl3,) d: 183,10 (CO); 163,00; 159,80; 155,30; 150,00; 138,90; 133,50; 131,60; 129,30; 127,20; 120,00; 117,30; 112,00; 95,60; 88,20; 58,90 (OCH3). EMAR m/z (M+) calculada para C17H13O4Br: 359,9997; encontrado 359,9958.
RESULTADOS E DISCUSSÃO
Síntese das a-bromo-acetofenonas
O nosso grupo de pesquisa tem atuado nos últimos cinco anos na síntese de produtos estratégicos com aplicação econômica e social16-26. O desenvolvimento de reações com este mesmo perfil, neste trabalho, é demonstrado pela investigação exaustiva de métodos eficientes para obtenção de a-bromo-acetofenonas a partir de acetofenonas e sua aplicação na síntese total das 2-benzoil-benzofuranas. Substâncias desta classe são utilizadas como anti-arrítmicos27, anti-adrenérgicos28 e aumentam a fibrilação ventricular em porcos apresentando miocardia isquêmica29.
As a-bromo-acetofenonas aromáticas substituídas são compostos de grande valor sintético, sendo amplamente empregadas como precursores na síntese de b-agonistas e de 2-benzoil-benzofuranas, substâncias que apresentam atividades biológicas importantes, tais como indução de hipertrofia muscular esquelética e vasodilatação coronariana28,29.
A partir de uma análise sintética prévia observando a estrutura química das 2-benzoil-benzofuranas, pudemos imaginar que as mesmas poderiam ser obtidas a partir dos orto-hidroxibenzaldeídos (8a-f) e das a-bromo-acetofenonas (4a-f), mediante uma reação de o-alquilação, seguida de uma condensação intramolecular.
O comportamento cromatográfico dos compostos (4) e de seu produto lateral (7), geralmente obtido nas bromações de acetofenonas, dificulta a sua purificação, o que nos levou a investigar metodologias viáveis, que favorecessem a formação dos compostos monobromados (4). Em nossa primeira abordagem, utilizamos como agente de bromação o Br2 em éter etílico (Esquema 1), um método clássico de obtenção de a-bromo-acetofenonas30. No entanto, esta reação não se apresentou satisfatória aos nossos objetivos, não só em função do baixo rendimento do produto desejado 4 (a-monobromado), como também pela formação dos subprodutos a,a-dibromados (7a, d, e) em quantidade significativa (Tabela 1). A formação de espécies dibromadas 7 deve-se à reação do Br2 com as espécies monobromadas 4, visto que estas ainda apresentam hidrogênios a-carbonílicos, podendo formar enóis, espécies passíveis de sofrerem uma nova reação de halogenação.
Em nossa segunda abordagem17 (Esquema 2), acreditando-se que condições mais brandas diminuiriam a probabilidade de se formar a espécie dibromada, optamos pelo emprego de CuBr2/ EtOAc/ CHCl3. Os produtos monobromados 4a, b, d, f foram obtidos em rendimentos satisfatórios e promissores, entretanto a presença de adutos dibromados também foi observada em pequenas quantidades (Tabela 1).
A aplicação do reagente tribrometo de feniltrimetilamônio em reações de bromação foi investigada por Marquet e Jacques31. Sua vantagem nestas reações tem sido atribuída a sua alta estabilidade, fácil preparo, além de uma eletrofilicidade menor e, portanto, maior seletividade, em relação as abordagens que utilizam átomos de bromo livres.
Desta forma, testamos a reação de a-bromocetalização (Esquema 3), empregando tribrometo de feniltrimentilamônio e etilenoglicol, conforme investigada por Visweswariah e colaboradores32 para cetonas alifáticas. Esta metodologia foi adaptada ao nosso sistema, fornecendo a-bromo-acetofenonas cetalizadas. Esta bromocetalização provavelmente envolve na primeira etapa a bromação do enol, o qual está em equilíbrio com a respectiva cetona, seguida de uma reação de cetalização com etilenoglicol, catalisada por HBr. A hidrólise posterior dos cetais durante o isolamento da reação forneceu em bons rendimentos as a-bromo-acetofenonas desejadas, praticamente isentas dos respectivos contaminantes a,a-dibromados 7a-f, após a observação dos espectros de RMN1H destas reações (Tabela 1). Com estes resultados ficou demonstrado que esta última metodologia de síntese de bromação é a mais recomendada para obtenção de produtos monobromados na posição a-metila da carbonila aromática das acetofenonas (6a-f).
Síntese das 2-benzoil-benzofuranas
O posterior tratamento das a-bromo-acetofenonas (4a-f) sintetizadas com o 2-hidroxi-benzaldeído apropriado (8a-f) em acetona, seca com K2CO3, sob refluxo por 4 h, resultou nas benzoil-benzofuranas (5a-s) (Esquema 4), através de uma ciclização intramolecular. Os rendimentos obtidos assim como o grau de pureza apresentaram-se satisfatórios, conforme indicado na Tabela 2.
O mecanismo proposto para a formação do anel benzofurânico (Esquema 5), a partir da reação de alquilação entre 4 e 8, consiste de 3 etapas. Na primeira (5.a), acreditamos que o tratamento do orto-hidroxibenzaldeído (8) com K2CO3 levará à formação do fenolato (9). Na seqüência, (5.b), o fenolato intermediário (9) desloca o halogênio da a-halocetona (4), formando o intermediário (10). Finalmente em (5.c), observamos a remoção do próton vicinal à carbonila e subseqüente condensação, formando o sistema benzoilbenzofurânico (5).
Com esta abordagem, as benzoil-benzofuranas foram obtidas em excelentes rendimentos, que não parecem ser afetados pela substituição do anel aromático em várias posições.
CONCLUSÃO
Uma nova metodologia para a síntese de a-bromo-acetofenonas (4a-f), e a aplicação destas na obtenção de 2-benzoil-benzofuranas (5a-s), através da reação de o-alquilação com 2-hidroxibenzaldeídos substituídos (8a-f) foi detalhadamente apresentada em excelentes rendimentos.
Diante da importância química e farmacológica desses compostos, e da baixa eficiência das demais metodologias em relação à regiosseletividade destes sistemas, explorando uma reação quimiosseletiva de bromação alifática em várias acetofenonas, a nova rota sintética desenvolvida abre um caminho prático e versátil de preparação destas substâncias.
AGRADECIMENTOS
À CAPES, CNPq, FAPERJ e FUJB pelo apoio financeiro e bolsas de estudo concedidas e às Centrais Analíticas IQ/UFRJ, NPPN- UFRJ e INCQS- FIOCRUZ.
REFERÊNCIAS
1. Bu'lock, J. D.; Hudson, A. T.; Kaye, B.; Chem. Commun.1967, 814.
2. Proksch, P.; Rodriguez, E.; Phytochemistry 1983, 22, 2335.
3. Basnet, P.; Kadota, S.; Terashima,S.; Shimizu, M.; Namba, T.; Chem. Pharm. Bull. 1993, 41, 1238.
4. Nomura, T.; Fukai,T.; Matsumoto, J.; Ohmori, T.; Planta Med. 1982, 46 , 28.
5. Kuo, G. Y.; Ross, S. T.; J. Heterocycl. Chem. 1978, 15, 1489.
6. Goldenberg, C.;Wandestrick, R.; Vanmeerbeeck, C.; Descamps, M.; Richard, J.; Bauthier, J.; Charlier, R.; Eur. J. Med. Chem. 1977, 12, 81; Mustafa, A.; Benzofurans, John Willey & Sons: New York, 1974.
7. Buu-Hoï, N. P. ; Bisagni, E.; Royer, R.; Routier, C. ; J. Chem. Soc. 1957, 625.
8. Singh, V. B.; Vaughan, W. E. M.; Chem. Abstr. 1970, 73, 97166.
9. Rap, E.; Beriche 1895, 26, 2901.
10. Dai, W-M.; Lai, K. W.; Tetrahedron Lett. 2002, 43, 9377.
11. Ono, M.; Kung, M-P.; Hou, C.; Kung, H. F.; Nucl. Med. Biol. 2002, 29, 633.
12. Smith, R. A.; Chen, J.; Mader, M. M.; Muegge, I.; Moehler, U.; Katti, S.; Marrero, D.; Stirtan, W. G.; Weaver, D. R.; Xiao, H.; Carley, W.; Bioorg. Med. Chem Lett. 2002, 12, 2875.
13. Perrin, D. D.; Armarego, W. L.; Purification of Laboratory Chemicals, 3rd ed., Butterworth-Heinemann Ltd.: Londres, 1988.
14. Still, W. C.; Kahn, M.; Mitra, A.; J. Org. Chem. 1978, 43, 2923.
15. Krüger, G.; Keck, J.; Noll, K.; Pieper, H.; Arzneim.- Forsch./Drug Res. 1984, 34, 1612.
16. Lopes, C. C.; Carvalho, J. R. M.; Cardoso, J. N.; Miranda, M. G.; Alves, G. B. C.; Lopes, R. S. C.; Ciência Hoje 2005, 36, 44.
17. Azevedo, M. S.; Alves, G. B. C.; Cardoso, J. N.; Lopes, C. C.; Lopes, R. S. C.; Synthesis 2004, 1262.
18. Lopes, C. C.; Lopes, R. S. C.; Mazzei, A. L. A.; Cardoso, J. N.; Silva, J. C.; Br PI 0400467-1, 2004.
19. Lopes, C. C.; Lopes, R. S. C.; Miranda, M. G.; Carvalho; J. R. M.; Cardoso, J. N.; Alves, G. B. C.; Br PI 0404130-5. 2004
20. Lopes, C. C.; Pereira, L. G.; Silva, J. A.; Cardoso J. N.; Lopes, R. S. C.; World Intellectual Property Office- Patent Cooperation Treaty, New York- USA, 2004.
21. Santos, R. P.; Amorim, M. B.; Lopes, C. C.; Lopes, R. S. C.; Quim. Nova 2003, 26, 216.
22. Nery, M. S.; Ribeiro, R. P.; Lopes, C. C.; Lopes, R. S. C.; Synthesis 2003, 272.
23. Da Silva, J. A.; Felcman, J.; Merce, A. L. R.; Mangrich, A. S.; Lopes, C. C.; .Lopes, R. S. C.; Inorg. Chim. Acta 2003, 356, 155.
24. Lopes, C. C.; Silva, J. A.; Cardoso, J. N.; Pereira, L. G.; Lopes, R. S. C.; Br PI 0307864-7, 2003
25. Lopes, C. C.; Silva, J. A.; Felcman, J.; Lopes, R S. C.; Villar, J. D. F.; Spectrosc. Lett. 2002, 35, 643.
26. Santos, R. P.; Lopes, C. C.; Lopes, R, S. C.; Synthesis 2001, 845.
27. Ha, H. R.; Follath, F.; Curr. Drug Metab. 2004, 5, 543.
28. Gautier, P.; Guillemare, E.; Djandjighian, L.; Marion, A.; Planchenaut, J.; Bernhart, C.; Herbert, J. M.; Nisato, D.; J. Cardiovasc. Pharmacol. 2004, 44, 244.
29. Tsagalou, E. P.; Anastasiou-Nana, M. T.; Charitos, C. E.; Siafakas, C. X.; Drakos, S. G.; Ntalianis, A.; Terrovitis, J. V.; Mavrikakis, E. M.; Doufas, A.; Nanas, J. N.; Resuscitation 2004, 61, 83.
30. March, J.; Advanced Organic Chemistry Reactions, Mechanisms and Structure, 5th Ed., John Willey & Sons: New York, 2001.
31. Marquet, A.; Jacques, J.; Tetrahedron Lett. 1959, 9, 24.
32. Visweswariah, S.; Prakash, G.; Bhushan, V.; Chandrasekaran, S.; Synthesis 1982, 309.
Recebido em 9/1/06; aceito em 30/3/06; publicado na web em 30/8/06
- 1. Bu'lock, J. D.; Hudson, A. T.; Kaye, B.; Chem. Commun.1967, 814.
- 2. Proksch, P.; Rodriguez, E.; Phytochemistry 1983, 22, 2335.
- 3. Basnet, P.; Kadota, S.; Terashima,S.; Shimizu, M.; Namba, T.; Chem. Pharm. Bull. 1993, 41, 1238.
- 4. Nomura, T.; Fukai,T.; Matsumoto, J.; Ohmori, T.; Planta Med. 1982, 46 , 28.
- 5. Kuo, G. Y.; Ross, S. T.; J. Heterocycl. Chem. 1978, 15, 1489.
- 6. Goldenberg, C.;Wandestrick, R.; Vanmeerbeeck, C.; Descamps, M.; Richard, J.; Bauthier, J.; Charlier, R.; Eur. J. Med. Chem. 1977, 12, 81;
- Mustafa, A.; Benzofurans, John Willey & Sons: New York, 1974.
- 7. Buu-Hoï, N. P. ; Bisagni, E.; Royer, R.; Routier, C. ; J. Chem. Soc. 1957, 625.
- 8. Singh, V. B.; Vaughan, W. E. M.; Chem. Abstr. 1970, 73, 97166.
- 9. Rap, E.; Beriche 1895, 26, 2901.
- 10. Dai, W-M.; Lai, K. W.; Tetrahedron Lett. 2002, 43, 9377.
- 11. Ono, M.; Kung, M-P.; Hou, C.; Kung, H. F.; Nucl. Med. Biol. 2002, 29, 633.
- 12. Smith, R. A.; Chen, J.; Mader, M. M.; Muegge, I.; Moehler, U.; Katti, S.; Marrero, D.; Stirtan, W. G.; Weaver, D. R.; Xiao, H.; Carley, W.; Bioorg. Med. Chem Lett. 2002, 12, 2875.
- 13. Perrin, D. D.; Armarego, W. L.; Purification of Laboratory Chemicals, 3rd ed., Butterworth-Heinemann Ltd.: Londres, 1988.
- 14. Still, W. C.; Kahn, M.; Mitra, A.; J. Org. Chem. 1978, 43, 2923.
- 15. Krüger, G.; Keck, J.; Noll, K.; Pieper, H.; Arzneim.- Forsch./Drug Res. 1984, 34, 1612.
- 16. Lopes, C. C.; Carvalho, J. R. M.; Cardoso, J. N.; Miranda, M. G.; Alves, G. B. C.; Lopes, R. S. C.; Ciência Hoje 2005, 36, 44.
- 17. Azevedo, M. S.; Alves, G. B. C.; Cardoso, J. N.; Lopes, C. C.; Lopes, R. S. C.; Synthesis 2004, 1262.
- 18. Lopes, C. C.; Lopes, R. S. C.; Mazzei, A. L. A.; Cardoso, J. N.; Silva, J. C.; Br PI 0400467-1, 2004
- 19. Lopes, C. C.; Lopes, R. S. C.; Miranda, M. G.; Carvalho; J. R. M.; Cardoso, J. N.; Alves, G. B. C.; Br PI 0404130-5. 2004
- 20. Lopes, C. C.; Pereira, L. G.; Silva, J. A.; Cardoso J. N.; Lopes, R. S. C.; World Intellectual Property Office- Patent Cooperation Treaty, New York- USA, 2004
- 21. Santos, R. P.; Amorim, M. B.; Lopes, C. C.; Lopes, R. S. C.; Quim. Nova 2003, 26, 216.
- 22. Nery, M. S.; Ribeiro, R. P.; Lopes, C. C.; Lopes, R. S. C.; Synthesis 2003, 272.
- 23. Da Silva, J. A.; Felcman, J.; Merce, A. L. R.; Mangrich, A. S.; Lopes, C. C.; .Lopes, R. S. C.; Inorg. Chim. Acta 2003, 356, 155.
- 24. Lopes, C. C.; Silva, J. A.; Cardoso, J. N.; Pereira, L. G.; Lopes, R. S. C.; Br PI 0307864-7, 2003
- 25. Lopes, C. C.; Silva, J. A.; Felcman, J.; Lopes, R S. C.; Villar, J. D. F.; Spectrosc. Lett. 2002, 35, 643.
- 26. Santos, R. P.; Lopes, C. C.; Lopes, R, S. C.; Synthesis 2001, 845.
- 27. Ha, H. R.; Follath, F.; Curr. Drug Metab. 2004, 5, 543.
- 28. Gautier, P.; Guillemare, E.; Djandjighian, L.; Marion, A.; Planchenaut, J.; Bernhart, C.; Herbert, J. M.; Nisato, D.; J. Cardiovasc. Pharmacol 2004, 44, 244.
- 29. Tsagalou, E. P.; Anastasiou-Nana, M. T.; Charitos, C. E.; Siafakas, C. X.; Drakos, S. G.; Ntalianis, A.; Terrovitis, J. V.; Mavrikakis, E. M.; Doufas, A.; Nanas, J. N.; Resuscitation 2004, 61, 83.
- 30. March, J.; Advanced Organic Chemistry Reactions, Mechanisms and Structure, 5th Ed., John Willey & Sons: New York, 2001.
- 31. Marquet, A.; Jacques, J.; Tetrahedron Lett. 1959, 9, 24.
- 32. Visweswariah, S.; Prakash, G.; Bhushan, V.; Chandrasekaran, S.; Synthesis 1982, 309.
Datas de Publicação
-
Publicação nesta coleção
06 Set 2011 -
Data do Fascículo
Dez 2006
Histórico
-
Recebido
09 Jan 2006 -
Aceito
30 Mar 2006