Resumo
In this work, we report the synthesis and the photoluminescence features of a Eu(III)-doped modified silica matrix obtained by the sol-gel method. The matrix was prepared by reaction between tetraethylorthosilicate and phenyltriethoxysilane alkoxide. The hydrolysis occurred using basic catalysis. The solids were treated at 100, 200 and 300 ºC during 4 h and the structure was determined by thermogravimetric analysis (TG/DTG), nuclear magnetic resonance (NMR 29Si and 13C), infrared spectroscopy (IR) and photoluminescence (PL). The PL spectra display the Eu(III) lines characteristic of the ion, 5D0 -> 7F J (J=0, 1, 2, 3, 4), the blue emission as ascribed in the silica matrix. The NMR and TG showed the stability of hybrid silica.
europium III; sol-gel; thermal analysis
europium III; sol-gel; thermal analysis
ARTIGO
Fenilsilicato dopado com Eu III obtido pelo método sol-gel
Phenylsilicate doped with Eu III obtained by sol-gel methodology
Eduardo J. Nassar* * e-mail: ejnassar@unifran.br ; Lilian R. Ávila; Paula F. S. Pereira; Evelisy C. O. Nassor; Alexandre Cestari; Katia J. Ciuffi; Paulo Sergio Calefi
Universidade de Franca, Av. Dr. Armando Salles Oliveira, 201, 14404-600 Franca SP, Brasil
ABSTRACT
In this work, we report the synthesis and the photoluminescence features of a Eu(III)-doped modified silica matrix obtained by the sol-gel method. The matrix was prepared by reaction between tetraethylorthosilicate and phenyltriethoxysilane alkoxide. The hydrolysis occurred using basic catalysis. The solids were treated at 100, 200 and 300 ºC during 4 h and the structure was determined by thermogravimetric analysis (TG/DTG), nuclear magnetic resonance (NMR 29Si and 13C), infrared spectroscopy (IR) and photoluminescence (PL). The PL spectra display the Eu(III) lines characteristic of the ion, 5D0® 7FJ (J=0, 1, 2, 3, 4), the blue emission as ascribed in the silica matrix. The NMR and TG showed the stability of hybrid silica.
Keywords: europium III; sol-gel; thermal analysis.
INTRODUÇÃO
Materiais híbridos obtidos pelo método sol-gel têm sido intensamente investigados nos últimos anos. As vantagens desta técnica baseiam-se na cinética de hidrólise e policondensação que permitem a interferência no sistema, através do pH, temperatura e solvente, oferecendo numerosas possibilidades de fabricação de amostras de sílica porosas e densas a baixas temperaturas e com morfologias controladas1. Um bom exemplo é a rota desenvolvida por Stöber, que consiste na preparação de partículas esféricas de sílica através da hidrólise e condensação do alcóxido tetraetilortossilicato, catalisado por uma base (NH4OH).
O processo sol-gel apresenta a vantagem de levar à formação de materiais híbridos mais homogêneos, além de ser possível, por esta metodologia, uma maior reprodutibilidade na obtenção dos materiais. As condições brandas envolvidas neste processo fornecem uma rota versátil para a preparação desses materiais2,3.
Os processos químicos básicos para formação de materiais obtidos pelo método sol-gel envolvem a hidrólise do precursor:
ºSi-OR + H2O ® ºSi-OH + ROH
e subseqüente formação da rede de silício:
ºSi-OH + HO-Siº ® ºSi-O-Siº + HOH
A habilidade para sintetizar polímeros inorgânicos usando o processo sol-gel com pouco ou nenhum aquecimento torna possível a dopagem destes géis com uma variedade de moléculas orgânicas e organometálicas, especialmente as que apresentam propriedades luminescentes4. Materiais óxidos, como a sílica, podem acomodar altas concentrações de dopantes adsorvidos em sua superfície, como os íons metálicos, mantendo uma boa homogeneidade, o que proporciona a formação de matrizes com importantes aplicações. Além disso, a superfície da sílica pode ser modificada com agentes sililantes, os quais podem ser adicionados simultaneamente à preparação da sílica ou, posteriormente, às suas sínteses5,6.
O processo sol-gel permite também a incorporação de redes de polímeros orgânicos, que levam à formação de materiais híbridos orgânico-inorgânicos7.
O conceito de nanocompósitos híbridos orgânico-inorgânicos explodiu nos anos 80 com a expansão dos processos químicos inorgânicos mais brandos. Certamente a condição de síntese oferecida pelo processo sol-gel permitiu uma mistura de componentes inorgânicos e orgânicos em escala nanométrica. Desde então, o estudo dos nanocompósitos híbridos funcionais tornou-se um campo de investigação crescente em novos materiais que relacionam funções orgânicas e inorgânicas e altamente versáteis, oferecendo um amplo campo de possibilidades para elaboração de materiais em termos de processo e propriedades químicas e físicas8.
Os materiais híbridos orgânico-inorgânicos podem ser obtidos de três maneiras distintas2,3: a molécula orgânica é misturada com um componente inorgânico (silano) na fase líquida do processo sol-gel. Após a gelatinização, a molécula orgânica é encapsulada nos poros da matriz polimérica (sem ligação covalente entre os fragmentos orgânicos e a matriz); a molécula orgânica reage com um silano precursor do sol-gel e após a gelação ela é ligada quimicamente ao óxido inorgânico (com ligação covalente entre os fragmentos orgânicos e inorgânicos) e, a molécula orgânica é ligada à superfície da matriz sólida por ligações de hidrogênio ou covalente.
Materiais híbridos luminescentes contendo íons de terras raras ligados covalentemente à cadeia de sílica ou adsorvidos em sua superfície também têm sido obtidos pelo processo sol-gel9. Esses materiais podem ser facilmente preparados e/ou modificados através da variedade de combinações dos componentes disponíveis, conferindo diferentes contribuições às propriedades do material resultante, o que possibilita modificações nas propriedades mecânicas, controle da porosidade e ajuste no balanço hidrofílico/hidrofóbico. Essa grande versatilidade, associada às suas propriedades ópticas, estabilidade química e termo-mecânica, faz com que esses materiais se apresentem com um grande potencial em diferentes aplicações, como no desenvolvimento de sensores químicos, aplicações ópticas, biomateriais, em catálise, revestimentos, aplicações cromatográficas, preparação de membranas e materiais compósitos10.
Neste trabalho, relatamos a síntese e as caracterizações de matrizes híbridas orgânico-inorgânicas à base de silício obtidas pela rota sol-gel convencional. A partir dos alcóxidos feniltrietoxisilano (PTES) e tetraetilortossilicato (TEOS), foram sintetizadas matrizes híbridas de sílicas orgânico-inorgânicas, utilizando-se a catálise básica. O cloreto de európio III foi utilizado como sonda estrutural e esses materiais foram caracterizados através da análise térmica (TG/DTG), espectroscopia no infravermelho (IV), ressonância magnética nuclear de silício e carbono (RMN 29Si e 13C) e fotoluminescência (FL) do íon Eu III.
PARTE EXPERIMENTAL
Preparação das sílicas
As sílicas foram preparadas através do processo sol-gel, pela adição do alcóxido TEOS (Aldrich), em etanol contendo PTES (Aldrich) na razão molar de 1:1 e 0:1. Adicionou-se o catalisador (NH4OH) concentrado até a completa precipitação da sílica. Este procedimento foi realizado por agitação vigorosa magnética por 30 min. O sólido formado foi filtrado, lavado com álcool etílico e seco na estufa à 50 ºC por 24 h. As amostras sofreram tratamento térmico por 4 h, nas temperaturas de 100, 200 e 300 ºC.
Caracterização
Análise térmica (TG/DTG)
As análises térmicas foram realizadas no aparelho TA Instruments- SDT Q600 -Simultaneous DTA-TG utilizando um gradiente de temperatura entre a temperatura ambiente (~25 ºC) até 1500 ºC, com uma taxa de aquecimento de 20 ºC/min e com o fluxo de nitrogênio em 100 mL/min.
Espectroscopia no infravermelho (IV)
Os espectros de infravermelho foram obtidos em espectrofotômetro Nicolet Protege 460, calibrado com filme e poliestireno (1601,4 cm-1). As amostras foram analisadas na forma de pastilhas de KBr.
Ressonância magnética nuclear de 29Si e 13C
Os resultados da ressonância magnética nuclear de 29Si e 13C RMN, em estado sólido, foram obtidos em um aparelho Espectrômetro Varian- INOVA 300 (59,5 MHz), usando como referência o rotor de nitrito de silício.
Fotoluminescência (FL)
Obtiveram-se os espectros de excitação e emissão em um espectrofluorimetro SPEX fluorolog F212l com lâmpada de xenônio contínua de 450 W como fonte de radiação, monocromador duplo SPEX modelo 1680 e fotomultiplicadora R 928 Hammatsu. As medidas foram coletadas a 22,5º em relação ao feixe incidente.
RESULTADOS E DISCUSSÃO
Termogravimetria
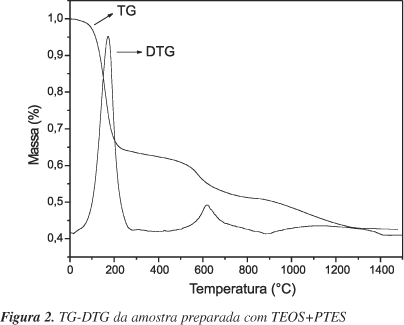
As Figuras 1 e 2 apresentam as curvas termogravimétricas e suas derivadas para as amostras preparadas a partir do precursor PTES e TEOS+PTES, respectivamente.
As análises térmicas das amostras apresentam curvas termogravimétricas (TG) bem distintas. A amostra obtida através da utilização do TEOS+PTES apresentou perda de massa de 36%; já para o PTES a perda de massa é de 3%. A perda de massa ocorre principalmente entre 100 e 200 ºC. Observa-se que ao se adicionar o precursor TEOS há um aumento na quantidade de água adsorvida; isto pode estar relacionado à presença de grupos OH na superfície da sílica, devido ao baixo grau de hidrólise e condensação dos grupos alcóxidos e, conseqüentemente, a um aumento na quantidade de água adsorvida. A diferença na quantidade de água pode ser melhor visualizada pela derivada da curva termogravimétrica (DTG), pois, de acordo com a literatura11, a área das DTG é diretamente proporcional às perdas de massa. Verificou-se também uma perda de massa na região de 400 a 800 ºC, atribuída à pirólise do substituinte orgânico do precursor PTES (grupo fenil).
A adição de TEOS provoca uma diluição da amostra; a perda de massa total para a amostra com PTES é em torno de 40%, sendo 3% relacionado à água e 37% à matéria orgânica. Já a amostra contendo TEOS+PTES apresenta uma perda total de 71%, sendo 36% de água e 35% da matéria orgânica. Cálculos teóricos demonstraram que na preparação da sílica modificada com TEOS e PTES, na proporção de 1:1 em mol, apresentam-se aproximadamente 38% de matéria orgânica, valor próximo ao obtido experimentalmente.
Com base nas curvas termogravimétricas, verifica-se que a estabilidade do grupo orgânico (fenil) foi até 400 ºC para as amostras TEOS+PTES e, para as amostras preparadas apenas com PTES, mostrou uma perda de massa constante, como se pode ver nas Figuras 1 e 2.
As amostras tratadas nas temperaturas de 100, 200 e 300 ºC apresentaram curvas termogravimétricas semelhantes, diferenciando somente nas perdas de massa relativas à quantidade de água.
Espectroscopia no infravermelho (IV)
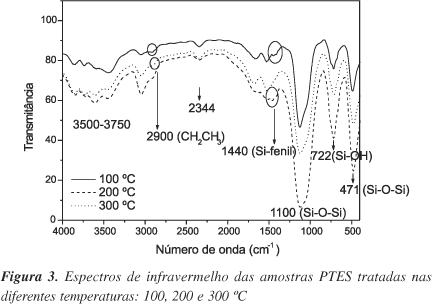
As Figuras 3 e 4 apresentam os espectros de IV das amostras preparadas somente com o precursor PTES e TEOS+PTES, tratadas nas diferentes temperaturas 100, 200 e 300 ºC.
Verificou-se uma pequena diferença nos espectros em relação ao número e à intensidade das bandas. Os espectros de infravermelho apresentam modos vibracionais da sílica em 470, 789 e 1100 cm-1 correspondentes às vibrações d(Si-O-Si), ns(Si-O-Si) e nas(Si-O-Si), respectivamente.
Vibrações originárias da absorção de moléculas de água na região de 3500-3700 cm-1 também foram observadas. Estas vibrações indicam a presença de grupos -OH na superfície da sílica e, também, a água remanescente da síntese, ratificando o que havia sido constatado na análise térmica: uma hidrólise e condensação incompleta.
Em 1440 cm-1 aparece a vibração correspondente à ligação Si-fenil, que aparece até 300 ºC para as amostras TEOS+PTES, e até 200 ºC para as amostras preparadas somente com o precursor PTES, indicando o início da decomposição do material orgânico. Este fato é confirmado pela análise térmica, que apresentou um início da decomposição da matéria orgânica a 400 e 200 ºC para as amostras contendo TEOS+PTES e PTES, respectivamente.
A presença da matéria orgânica também é confirmada com a banda em 2900 cm-1, correspondente às vibrações C-H.
SegundoVinod et. al. 12, em polímeros organossilânicos, a banda observada em 2350 cm-1 é atribuída a defeitos na superfície da sílica, banda observada para todas as amostras, independente da temperatura a que foram submetidas.
RMN 29Si
A utilização da ressonância magnética nuclear (RMN) é uma técnica bastante útil para determinação de estruturas de materiais preparados por essa metodologia. O grau de condensação destes materiais pode ser medido através da porcentagem de grupos Qn e Tn13. A Tabela 1 apresenta as posições dos picos no espectro de ressonância magnética nuclear do silício nas amostras.
A amostra PTES, seca à temperatura ambiente (TA), apresentou apenas um deslocamento químico em -66 ppm, devido ao silício ligado ao carbono do grupo fenil e teve as outras posições hidrolisadas. A amostra contendo TEOS+PTES (TA) apresentou hidrólise e condensação incompleta, indicada pela presença dos picos relativos aos deslocamentos T2 e Q3, que também foi observado para a amostra tratada a 100 ºC. Para a amostra tratada a 200 ºC, o pico referente ao carbono T2 desaparece, provavelmente devido à transformação deste em T3. Já na amostra aquecida a 300 ºC, o pico Q4 aparece mais intenso que os demais e indica um aumento na condensação do silício. Isto mostra uma dependência do grau de condensação em relação à temperatura de tratamento das amostras.
RMN 13C
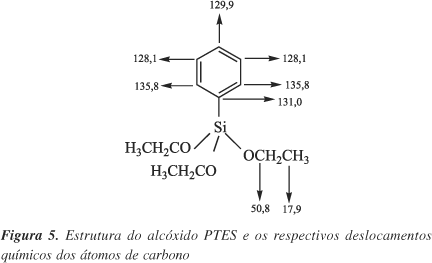
A Figura 5 apresenta a molécula do alcóxido PTES com os deslocamentos químicos simulados por um programa computacional.
Nota-se que são esperados 6 sinais no espectro de RMN de 13C para as amostras contendo o PTES. Na Figura 6 é apresentado o espectro de RMN de 13C para a amostra contendo somente PTES após ter sofrido hidrólise (pó).
Pode-se observar no espectro o aparecimento de picos correspondendo aos deslocamentos 18, 50, 128, 130, 131 e 135 ppm, com pequenas diferenças em relação ao previsto teoricamente. Os deslocamentos 18 e 50 ppm foram atribuídos aos carbonos do grupo etoxi, originários dos alcóxidos TEOS e PTES. Esses sinais desaparecem nas temperaturas de 200 e 300 ºC, indicando a eliminação desses grupos e comprovando o aumento do grau de hidrólise e condensação observado nos RMN 29Si .
Os outros sinais 128, 130, 131 e 135 ppm referentes ao grupo fenil também estão coerentes com o previsto. Nas temperaturas de 200 e 300 ºC ocorre uma diminuição na intensidade do pico em 131 ppm, correspondente à ligação Si-C; provavelmente o grupo orgânico não está mais ligado ao silício, pois, com o aumento da temperatura, o grupo fenil se decompõe, o que pode ser confirmado pelas curvas termogravimétricas.
O RMN de 29Si, para a amostra tratada a 300 ºC, mostrou que não se tem Si ligado ao C, porém o sinal de carbono ainda permanece. Portanto, pode-se dizer que a ligação Si-C pode ter sido rompida com o aumento da temperatura, mas que o fenil continua presente na sílica, provavelmente em seus poros, e a sofrer influência da vizinhança.
Fotoluminescência
A Figura 7 mostra os espectros de excitação do íon Eu III dopado na matriz de sílica, preparada com PTES e TEOS+PTES, monitorado em 612 nm (5D0® 7F2). As linhas são atribuídas às transições entre os níveis 7F0 (fundamental) e 5L6 e 5D1-3 (excitados).
Observa-se uma pequena diferença na região de maior energia com o máximo aparecendo em 276 nm, atribuída ao grupo fenil do precursor PTES.
Nas Figuras 8 e 9 são apresentados os espectros de excitação para as amostras preparadas com PTES e TEOS+PTES, tratadas termicamente.
Pode-se verificar uma mudança nos espectros de excitação do íon Eu III em função do aquecimento a que foram submetidas as amostras que apresentam a banda de excitação em 393 nm correspondente à transição 7F0® 5L6. A banda larga na região de 270 a 370 nm é diretamente dependente da temperatura.
Na Figura 10 são apresentados os espectros de emissão para as amostras preparadas com PTES e TEOS+PTES, excitadas em 394 nm (nível 5L6 do íon Eu III).
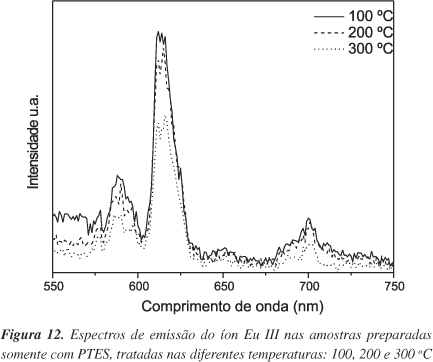
Nas Figuras 11 e 12 são mostrados os espectros de emissão para as amostras preparadas com PTES e TEOS+PTES, tratadas termicamente.
Pode-se notar as bandas correspondentes às transições do estado excitado 5D0 para o estado fundamental 7FJ (J = 0, 1, 2, 3 e 4). As bandas largas de emissão para as amostras obtidas à temperatura ambiente indicam uma ocupação não homogênea do íon Eu III na estrutura da sílica14-16. A presença da banda em 579 nm é relativa à transição 5D0® 7F0, indicando que o íon Eu III está ocupando sítios sem centro de inversão.
As amostras preparadas com TEOS+PTES tratadas termicamente apresentam uma banda larga de emissão na região azul do espectro eletromagnético. Alguns autores têm atribuído esta banda larga à própria sílica inorgânica17 e a grupos contendo nitrogênio presente em híbridos orgânico-inorgânicos18,19. No nosso caso, em particular, a presença do N poderia ter sua origem no catalisador utilizado, o NH4OH; porém, após aquecimento, todo hidróxido foi decomposto em amônia e liberado para o ambiente. Portanto, a banda larga de emissão na região azul do espectro eletromagnético pode ser atribuída à própria matriz de sílica.
O máximo de emissão da banda larga mostra ser dependente da temperatura de aquecimento a que foram submetidas as amostras: tratadas a 100, 200 e 300 ºC apresentaram máximo de emissão em 467, 477 e 484 nm, respectivamente. Observamos que, ao aumentarmos a temperatura de tratamento das amostras, aumenta-se o comprimento de onda, isto é, diminui a energia. Além da mudança no máximo de emissão, também observamos um alargamento na banda. A mudança de 10 e 7 nm da temperatura de 100 para 200 e de 200 para 300 ºC, respectivamente, pode inicar um aumento na incorporação dos íons nas dimensões do retículo inorgânico do silício17.
A banda correspondente à transição 5D0® 7 F1 é de natureza puramente dipolo-magnético e a sua intensidade é muito pouco afetada pelo ambiente do campo cristalino ao qual está submetido o íon Eu III. Assim, ela pode ser tomada como um padrão para medir as intensidades relativas das outras bandas do espectro20,21. As intensidades relativas são medidas em termos das Áreas Relativas, sob as bandas correspondentes às transições 5D0® 7F0 e 5D0® 7F2, em relação à banda correspondente à transição 5D0® 7F1. A Tabela 2 apresenta as Áreas Relativas (AR).
A diminuição da razão entre as AR correspondentes às transições 5D0® 7F2 / 5D0® 7F1 é um indicativo do aumento da simetria ao redor do íon Eu III em função da temperatura para as amostras contendo TEOS+PTES. Para as amostras tratadas termicamente a 100, 200 e 300 ºC contendo apenas PTES o ambiente do íon Eu III permaneceu o mesmo, pois a razão das AR não apresentou mudanças significativas.
CONCLUSÃO
A presente investigação demonstra a preparação de matrizes híbridas orgânico-inorgânicas pela rota sol-gel convencional. A adição do TEOS proporcionou um aumento na estabilidade térmica do material.
A presença do grupo fenil foi confirmada pelos espectros de infravermelho até as temperaturas de 400 e 200 ºC para as amostras TEOS+PTES e PTES, respectivamente.
A emissão do íon Eu III sofre influência do meio e algumas amostras estão relacionadas aos grupos OH da superfície da sílica, produzidos pela hidrólise e condensação incompleta dos grupos alcóxidos, como observado através dos espectros de RMN do 29Si. Esses grupos promovem perdas de energia por mecanismos vibracionais, despopulando o estado excitado 5D0.
A banda na região azul do espectro eletromagnético pode ser eliminada com o aumento da temperatura de tratamento das amostras, porém eliminaríamos também o grupo fenil, utilizado para agir como antena, absorvendo energia e transferindo-a para o íon Eu III; porém, é a presença desse grupo que pode proporcionar as novas propriedades para o material híbrido orgânico-inorgânico.
AGRADECIMENTOS
À FAPESP, ao CNPq e à CAPES pelos auxílios financeiro e bolsas concedidas. Ao Laboratório de Terras Raras da USP/Rib. Preto, pelas medidas de luminescência. Ao Prof. Dr. A. Federman Neto pela simulação computacional do RMN do percursor PTES.
REFERÊNCIAS
1. Nassar, E. J.; Messaddeq, Y.; Ribeiro, S. J. L.; Quim. Nova 2002, 25, 27.
2. Mark, J. E.; Lee, C. Y.C.; Bianconi, P. A.; Hybrid Organic-Inorganic Composites, ACS Symp. Ser. 586, American Chemical Society: Washington, DC, 1995.
3. Cerveau, G.; Corriu, R. J. P.; Lepeytre, C.; Mutin, P. H.; J. Mater. Chem. 1998, 8, 2707; Corriu, R. J. P.; Polyhedron 1998, 17, 925; Corriu, R. J. P.; Leclerq, D.; Angew. Chem., Int. Ed. 1996, 35, 1420; Shea, K. J.; Loy, D. A.; Webster, O.; J. Am. Chem. Soc. 1992, 114, 6700; Jackson, C. L.; Bauer, B. J.; Nakatami, A. I.; Barnes, J. D.; Chem. Mater. 1996, 8, 727.
4. Lobnik, A.; Majcen, N.; Niederreiter, K.; Uray, G.; Sens. Actuators, B 2001, 74, 200.
5. Gushiken, Y.; Moreira, J. C.; J. Colloid Interface Sci. 1985, 107, 70.
6. Nassar, E. J.; Ciuffi, K. J.; Ribeiro, S. J. L.; Messaddeq, Y.; Mater. Res. 2003, 6, 557.
7. Jitianu, A.; Gartner, M.; Zaharescu, M.; Cristea, D.; Manea, E.; Mater. Sci. Technol.; 2003, 23, 301.
8. Sanchez, C.; Julián, B.; Belleville, P.; Popall, M.; J. Mater. Chem. 2005, 15, 3559.
9. Klonkowski, A. M.; Mater. Sci. 2002, 20, 1.
10. José, N. M.; Prado, L. A. S.de A.; Quim. Nova 2005, 28, 281.
11. Mothé, C. G.; Azevedo, A. D.; Análise Térmica de Materiais, i Editora: São Paulo, 2002, p. 27-112.
12. Vinod, P. M.; Bahnemann, D.; Rajamonhanan, R. B.; Vijayamohanan, K.; J. Phys. Chem. B 2003, 107, 11583.
13. Kirk-Othmer-Encyclopedia of Chemical Technology, A. Wiley-Intersciense Publication- John Wiley & Sons, 4th ed.; vol. 16; Kirk-Othmer-Encyclopedia of Chemical Technology, A. Wiley-Intersciense Publication- John Wiley & Sons: New York, 4th ed.; vol. 14, p. 107.
14. Hazenkamp, M. F.; van der Veen, A. M. H.; Feiken, W.; Blasse, G.; J. Chem. Soc., Faraday Trans. 1992, 88, 141.
15. Rice, D. K.; Deshaser, L. G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1969, 186, 387.
16. Reisfeld, R.; J. Eletrochem. Soc. 1984, 131, 1360.
17. Carlos, L. D.; de Zea Bermudez, V.; Sá Ferreira, R. A.; J. Non-Cryst. Solids 1999, 247, 203.
18. Carlos, L. D.; Sá Ferreira, R. A.; de Zea Bermudez, V.; Ribeiro, S. J. L.; Adv. Funct. Mater. 2001, 11, 111.
19. Carlos, L. D.; Sá Ferreira, R. A.; Pereira, R. N.; Assunção, M.; de Zea Bermudez, V.; J. Phys. Chem. B 2004, 108, 14924.
20. Reisfeld, R.; Struct. Bonding 1979, 30, 65.
21. Blasse, G.; Solid State Chem. 1982, 3, 153.
Recebido em 14/7/06; aceito em 15/2/07; publicado na web em 30/7/07
- 1. Nassar, E. J.; Messaddeq, Y.; Ribeiro, S. J. L.; Quim. Nova 2002, 25, 27.
- 2. Mark, J. E.; Lee, C. Y.C.; Bianconi, P. A.; Hybrid Organic-Inorganic Composites, ACS Symp. Ser. 586, American Chemical Society: Washington, DC, 1995.
- 3. Cerveau, G.; Corriu, R. J. P.; Lepeytre, C.; Mutin, P. H.; J. Mater. Chem 1998, 8, 2707;
- Corriu, R. J. P.; Polyhedron 1998, 17, 925;
- Corriu, R. J. P.; Leclerq, D.; Angew. Chem., Int. Ed. 1996, 35, 1420;
- Shea, K. J.; Loy, D. A.; Webster, O.; J. Am. Chem. Soc. 1992, 114, 6700;
- Jackson, C. L.; Bauer, B. J.; Nakatami, A. I.; Barnes, J. D.; Chem. Mater. 1996, 8, 727.
- 4. Lobnik, A.; Majcen, N.; Niederreiter, K.; Uray, G.; Sens. Actuators, B 2001, 74, 200.
- 5. Gushiken, Y.; Moreira, J. C.; J. Colloid Interface Sci. 1985, 107, 70.
- 6. Nassar, E. J.; Ciuffi, K. J.; Ribeiro, S. J. L.; Messaddeq, Y.; Mater. Res. 2003, 6, 557.
- 7. Jitianu, A.; Gartner, M.; Zaharescu, M.; Cristea, D.; Manea, E.; Mater. Sci. Technol.; 2003, 23, 301.
- 8. Sanchez, C.; Julián, B.; Belleville, P.; Popall, M.; J. Mater. Chem. 2005, 15, 3559.
- 9. Klonkowski, A. M.; Mater. Sci. 2002, 20, 1.
- 10. José, N. M.; Prado, L. A. S.de A.; Quim. Nova 2005, 28, 281.
- 11. Mothé, C. G.; Azevedo, A. D.; Análise Térmica de Materiais, i Editora: São Paulo, 2002, p. 27-112.
- 12. Vinod, P. M.; Bahnemann, D.; Rajamonhanan, R. B.; Vijayamohanan, K.; J. Phys. Chem. B 2003, 107, 11583.
- 13. Kirk-Othmer-Encyclopedia of Chemical Technology, A. Wiley-Intersciense Publication- John Wiley & Sons, 4th ed.; vol. 16;
- Kirk-Othmer-Encyclopedia of Chemical Technology, A. Wiley-Intersciense Publication- John Wiley & Sons: New York, 4th ed.; vol. 14, p. 107.
- 14. Hazenkamp, M. F.; van der Veen, A. M. H.; Feiken, W.; Blasse, G.; J. Chem. Soc., Faraday Trans. 1992, 88, 141.
- 15. Rice, D. K.; Deshaser, L. G.; Phys. Rev. B: Condens. Matter Mater. Phys. 1969, 186, 387.
- 16. Reisfeld, R.; J. Eletrochem. Soc. 1984, 131, 1360.
- 17. Carlos, L. D.; de Zea Bermudez, V.; Sá Ferreira, R. A.; J. Non-Cryst. Solids 1999, 247, 203.
- 18. Carlos, L. D.; Sá Ferreira, R. A.; de Zea Bermudez, V.; Ribeiro, S. J. L.; Adv. Funct. Mater. 2001, 11, 111.
- 19. Carlos, L. D.; Sá Ferreira, R. A.; Pereira, R. N.; Assunção, M.; de Zea Bermudez, V.; J. Phys. Chem. B 2004, 108, 14924.
- 20. Reisfeld, R.; Struct. Bonding 1979, 30, 65.
- 21. Blasse, G.; Solid State Chem. 1982, 3, 153.
Datas de Publicação
-
Publicação nesta coleção
11 Dez 2007 -
Data do Fascículo
2007
Histórico
-
Aceito
15 Fev 2007 -
Recebido
14 Jul 2006