
Resumo
This review reports the application of inorganic and organic polymeric materials for cation removal by using nitrogenated basic centers. The data demonstrate the importance of the desired groups when free or immobilized on natural or synthesized inorganic polymers through silanol groups. Thus, the most studied silica gel is followed by natural crysotile and talc polymers, and the synthesized mesopore silicas, talc-like, silicic acids, phosphates and phyllosilicates. The organic natural biopolymeric chitin and cellulose were chemically modified to improve the availability of the amine groups or the reactivity with desirable molecules to enlarge the content of basic centers. The cation removal takes place at the solid/liquid interface and some interactive effects have their thermodynamic data determined.
inorganic polymer; biopolymer; cation removal
inorganic polymer; biopolymer; cation removal
REVISÃO
A relevante potencialidade dos centros básicos nitrogenados disponíveis em polímeros inorgânicos e biopolímeros na remoção catiônica
The weighty potentiality of nitrogenated basic centers in inorganic polymers and biopolymers for cation removal
Claudio Airoldi* * e-mail: airoldi@iqm.unicamp.br
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas - SP, Brasil
ABSTRACT
This review reports the application of inorganic and organic polymeric materials for cation removal by using nitrogenated basic centers. The data demonstrate the importance of the desired groups when free or immobilized on natural or synthesized inorganic polymers through silanol groups. Thus, the most studied silica gel is followed by natural crysotile and talc polymers, and the synthesized mesopore silicas, talc-like, silicic acids, phosphates and phyllosilicates. The organic natural biopolymeric chitin and cellulose were chemically modified to improve the availability of the amine groups or the reactivity with desirable molecules to enlarge the content of basic centers. The cation removal takes place at the solid/liquid interface and some interactive effects have their thermodynamic data determined.
Keywords: inorganic polymer; biopolymer; cation removal.
INTRODUÇÃO
Nos dias de hoje, quando se veicula assuntos relacionados com superfícies, principalmente na área de Química Inorgânica, os interessados se voltam mais diretamente à rotulação dos profissionais atuantes como químicos de materiais. Porém, muitas vezes são esquecidos os aspectos envolvendo as ligações efetuadas entre os átomos, que estão em quantidade maiores ou menores na formação do esqueleto polimérico. Mesmo sem o conhecimento de tais detalhes e ainda sobre a estrutura é possível empregar o material em atividades tanto acadêmicas como tecnológicas, sendo comumente destacada a propriedade da adsorção. Curiosamente, o material derivado da queima ou extraído de minas naturais, após tratamento, deriva o chamado carvão ativado, o qual foi o primeiro material usado para a remoção de metais pesados contidos em efluentes industriais. No entanto, em qualquer processo operacional, o custo envolvido precisa ser examinado para que seja viável o uso, pois o preço cresce em função das especificidades requeridas e, de fato, a purificação é bastante relevante, o que chega a inviabilizá-lo na adsorção, mas que possibilita o emprego mais amplo somente em pequenas escalas laboratoriais1.
Diante dos aspectos econômicos limitantes na obtenção do carvão de alta pureza, as pesquisas foram paulatinamente direcionadas para outras fontes alternativas. Um ponto marcante neste estudo foi buscar materiais naturais de baixo custo, que pudessem ter alta capacidade de adsorção, o que implicava na existência de centros quimicamente ativos na superfície, para que pudessem atuar com alta eficiência na retenção de cátions indesejáveis existentes em um leito aquoso contaminado. Nesta direção, o progresso do campo passou a ser mais eficaz após o conhecimento das funções químicas disponíveis nas superfícies dos materiais e possíveis tipos de ligações que poderiam formar frente a um determinado soluto, no decorrer do processo de adsorção na interface sólido/líquido2.
Uma maneira de se obter o material com baixo custo é explorar fontes amplamente encontradas na natureza, que exijam operações simples para chegar a um composto final de composição definida e de alta pureza, fato que nem sempre é alcançado. Dentre uma série de materiais, os zeólitos se ajustam com essas propriedades, o que levou a um grande interesse científico, principalmente por ser encontrado em depósitos naturais e pela alta capacidade de troca iônica. Esses fatos levaram muitas indústrias a se fixarem próximas às fontes extrativas, com grande vantagem e eficiência na remoção de cátions existentes em leitos fluidos contendo metais pesados. Identicamente, as argilas são versáteis nessas propriedades requeridas e também despontam como potencial alternativo ao carvão. Assim como os zeólitos, as argilas também são importantes componentes do solo. As argilas destacam-se pela habilidade em adsorver cátions devido à enorme área superficial e capacidade de troca, pois, a carga negativa encontrada na superfície facilita a formação de interações iônicas com os cátions disponíveis no meio2.
Em relação a outros possíveis materiais inorgânicos, sempre visando o custo, quase que invariavelmente eles são produtos secundários provenientes da indústria, normalmente produzidos em larga escala, portanto, passam a ser disponíveis a baixo custo. O emprego torna-se altamente desejável para evitar o acúmulo próximo à produção, cujo descarte necessita de várias operações, além da dificuldade em encontrar local apropriado, sem causar impacto ao meio ambiente. Nesta direção alguns materiais já foram testados nas possíveis etapas de remoção de metais pesados de água contaminada2, como cinzas, óxidos metálicos, sílica gel, argilas, lignina etc. Ainda no caso de carvão previamente oxidado, material encontrado em fontes naturais, contém as funções ácidas carboxílicas ou fenólicas, que facilitam a remoção dos cátions de ambientes aqüífero, solo etc. Porém, em muitas oportunidades o simples uso desses materiais secundários tal como produzidos ou encontrados na natureza são estudados como alternativos, mas que em muitos casos são abandonados devido ao baixo poder na adsorção1,2.
Em se tratando de constituições poliméricas derivadas de biopolímeros, alguns polissacarídeos merecem atenção quanto ao próprio material in natura ou extraído através de processos químicos, para que atenda às mesmas características de remoção de metais e sejam comparáveis ao carvão ativado. Dentre esses, os mais explorados são a celulose e a quitina, que são nessa ordem os mais abundantes biopolímeros encontrados na natureza, extraídos de vegetais e pequenos animais, respectivamente. No entanto, tal como obtidos possuem baixa capacidade de reação devido às ligações entre cadeias, impossibilitando a disponibilidade de grupos que potencialmente poderiam interagir com outros centros reativos. Para que o uso desses materiais seja aumentado é necessário que processos químicos possam viabilizar grupos reativos nas cadeias poliméricas e esta é a tendência natural para tornar aproveitáveis os recursos naturais3.
A utilidade de materiais de caráter polimérico, tanto inorgânico como orgânico, está intimamente relacionada com o aumento de descarga dos componentes metálicos em um determinado sistema, pois qualquer fração adicional ao meio, além da inerente composição natural, causa desequilíbrio ambiental à flora e fauna, além da possibilidade de atingir o lençol freático para refletir em distúrbios ao homem. Desta forma, em defesa da manutenção das condições ambientais vários segmentos da sociedade têm-se manifestado, estando ou não diretamente envolvidos com a Química, o que aumenta a responsabilidade desse profissional em evitar e prontamente agir para estabelecer as condições mais adequadas, para buscar e propor o uso de materiais que sejam aplicáveis em soluções dos problemas ambientais. Como resultado, as investigações direcionadas a esse mister são sempre bem acolhidas pela comunidade, principalmente quando novos materiais são propostos para tal finalidade, como será aqui apresentado com destaque na potencialidade do centro básico nitrogenado disponível, ou inseridos através de processos sintéticos nas estruturas poliméricas.
POLÍMEROS INORGÂNICOS
Esses polímeros apresentam originalmente ou existem condições para que possam expor grupos hidroxilas em suas superfícies, cuja polaridade natural entre os átomos constituintes possibilita a formação de ligações de hidrogênio. Assim, quaisquer superfícies poliméricas (º|) contêm moléculas de água ligadas diretamente ao grupo hidroxila (º|-OH), que por sua vez estão ligadas a outras idênticas moléculas, acumulando camadas sucessivas, as quais são eliminadas sob aquecimento controlado, para que se obtenham as melhores condições de reação do grupo livre, que é a fonte reativa da superfície4. Nessas condições, em presença de quaisquer moléculas básicas (: B), podem formar ligações de hidrogênio, Equação 1:

Essa interação quantitativa ácido-base pode ser perfeitamente mensurável através de titulações calorimétricas, como observado com a sílica gel, sendo que os valores entálpicos resultantes permitem estabelecer a ordem de basicidade dos vários reagentes envolvidos no processo, cujos resultados termodinâmicos permitem o entendimento dos efeitos interativos a respeito desse discutível e importante tipo de ligação, que afetam drasticamente as propriedades da maioria dos materiais5.
Vários materiais naturais, como, por exemplo, a própria argila, são encontrados carregados, isto é, apresentam metais nas superfícies externas, para contrabalançar as cargas existentes na constituição polimérica, normalmente na forma de cátions de metais alcalinos (M1+) que por sua vez em solução aquosa, podem ser trocáveis com outro cátion (M2+) também disperso em água, quando em condições de equilíbrio na interface sólido/líquido6, Equação 2:

Do ponto de vista reacional, o que mais se torna atrativo é explorar a reatividade dos grupos hidroxilas nas superfícies, pelo fato que o uso de reagentes adequados possa imobilizar moléculas ou, uma vez ligado à superfície inorgânica possa sofrer uma seqüência de reações, com o intuito de aumentar a cadeia pendente precursora. Os reagentes que se prestam a essa reação são os agentes sili-lantes, que possuem a fórmula geral, (X)3Si(R)Y, sendo X um grupo alcóxido ou halogênio, R é denominado de grupo espaçador, normalmente (CH2)3, e Y apresenta-se na forma mais variada, como Cl, NH2, CN, NCO, SH etc. Assim, uma vez imobilizado o agente sobre quaisquer superfícies, que consiste na denominada rota heterogênea, Equação 3, abre-se uma gama de possibilidades reacio-nais7-9. Por outro lado, as mesmas reações podem ser igualmente efetuadas em meio não aquoso, para se evitar hidrólise do agente sililante, dentro de uma rota denominada homogênea, Equação 4, antes da etapa de imobilização2,10.
Em ambos os casos o remanescente grupo X, após o enxerto, pode sofrer hidrólise na lavagem do material resultante com água, para dar novo grupo hidroxila, o qual complementa a ligação com a superfície.

Como se nota, o crescimento do campo depende da utilização dos mais variados suportes e basicamente das condições reacionais envolvendo os agentes sililantes, com suas respectivas capacidades de reação para aumentar a cadeia. No presente caso, será explorado o grupo amino, originário do agente sililante, que após reagir ainda manterá as características de centro básico, As superfícies poliméricas orgânicas também poderão dispor do mesmo grupo amino, que também é sensível à mesma sistemática reacional. Esse será o enfoque da presente exposição.
Em se tratando da imobilização em polímeros inorgânicos, a sílica gel amorfa é, sem dúvida, a mais estudada com uma variedade incrível em utilidades, pois a simples ligação covalente estabe-lecida na superfície do polímero faz com que desperte enorme interesse em várias áreas de pesquisa, como adsorção, catálise, separação e sensores, além da possibilidade de formar híbridos inorgânico-orgânico. Em particular, o grande avanço nesse campo provém do método reformador proveniente da formação de micelas, que possibilita a obtenção de sílicas mesoporosas. Essas apresentam enorme área superficial e poros, cujas propriedades facilitam a acomodação de moléculas no interior dos poros, em larga vantagem quando comparados com os materiais microporosos envolvidos na extensa Química hospedeiro-convidado11.
A reatividade tanto da sílica gel amorfa propriamente dita, como de outros polímeros contendo sílica em suas superfícies, está associada à alta concentração de grupos silanóis Si-OH, os quais são centros reativos que possibilitam a imobilização de moléculas, com o objetivo de modificar a superfície, como também no interior de poros. O que desperta muita atenção nos últimos anos é, sem dúvida, a modificação química de sílica mesoporosa, cujos enfoques são direcionados para aplicações ambientais, na remoção de cátions tóxicos através do processo de adsorção12,13.
Muito embora o princípio de imobilização seja idêntico do ponto de vista reacional, em quaisquer superfícies inorgânicas, aqui serão tratados em diferentes tópicos, ilustrando sobre a sílica gel amorfa, crisotila, filossilicato etc.
SÍLICA GEL
Os primeiros sucessos no sentido de imobilizar moléculas que pudessem ter grupos amino na superfície de sílica gel basearam-se no aproveitamento que surgiu após a síntese dos compostos clorados, derivados dos silanos. Vale acrescentar que a síntese e purificação desses compostos, inicialmente com interesse na eletrônica, eram por demais complexas e cercadas de cuidados especiais. No caso particular, para ilustrar as dificuldades existentes no preparo de precursores para a imobilização, a síntese efetuada em longo perío-do de trabalho, partiu-se de uma liga de ferro-silício, material extraído para exportação, que reagia com ácido clorídrico. A mistura obtida era cuidadosamente destilada e o produto de interesse isolado reagia com cloreto de alila, numa reação catalisada por platina. Após a destilação o composto isolado, Equação 5, era tratado com álcool para formar o respectivo alcóxido e em seguida com a molécula de interesse, etilenodiamina14, como mostra a Equação 6, na obtenção do agente sililante.

Como houve grande interesse nesses tipos de agentes sililantes, os produtos comerciais passaram a ser encontrados, principalmente o precursor 3-aminotrialcoxissilano, o que facilitou o avanço do campo. Em quaisquer imobilizações, sendo mais facilmente efetivado pela rota heterogênea, o processo consiste em suspender a sílica gel em condições anidras, com adição do agente de interesse, por longo tempo de refluxo na temperatura de ebulição do sol-vente apolar empregado, para fixar a molécula de maneira covalente na superfície de sílica gel.
Em todas as imobilizações o que se constata é a permanência de um grupo alcóxido aparentemente inerte, como mostram as reações 3 e 4, mas que na lavagem sofre hidrólise e possivelmente interage com os remanescentes grupos silanóis da superfície, e, portando, para simplicidade não mais será apresentado nas ilustrações. Para simplificar em todas as imobilizações, no produto final de reação, apenas será representada a molécula ligada cova-lentemente na superfície, como mostra a Equação 7.
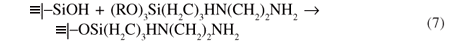
A potencialidade em aumentar a cadeia do agente sililante pode ser constatada através de um dialdeído, cuja reação controlada em termos estequiométricos, superfície aminada: glutaraldeído, forma facilmente a base de Schiff, a qual pode ser reduzida em etapas posteriores, para seguir uma seqüência de reações para conduzir ao produto final com claro crescimento da cadeia15, como mostra a Equação 8. Após a imobilização e redução da dupla ligação, o material possui a capacidade de remover cátions16. Da mesma maneira, os compostos aminados são úteis na extração seletiva de metais do grupo da platina, existentes em efluentes industriais de refinaria17.

O agente sililante 3-cloropropilalcoxissilano é também um precursor altamente utilizado para imobilizar moléculas na superfície da sílica, por exemplo, como acontece na incorporação da molécula 2-aminometilpiridina, cuja imobilização tem como objetivo aumentar os centros básicos para a melhor efetivação da adsorção18, como mostra a Equação 9. O mesmo precursor quando incorpora a molécula de dietilenotretamina facilita a subseqüente reação com mono e bis-naftaldeído e salicilaldeído, através do favorecimento da formação da base de Schiff. Num processo inverso os mesmos aldeídos reagiram previamente com a molécula de dietilenotetramina, cujo produto de reação foi imobilizado no suporte precursor19. A mesma reação de imobilização foi verificada com o grupo amino do precursor ou mesmo quando a molécula de etilenodiamina foi previamente ancorada, seguida da reação com derivados do ácido formilsalicílico20. A inclusão de átomos de nitrogênio em cadeia cíclica de pirazina também dá flexibilidade às moléculas pendentes ligadas covalentemente na estrutura polimérica da sílica21.

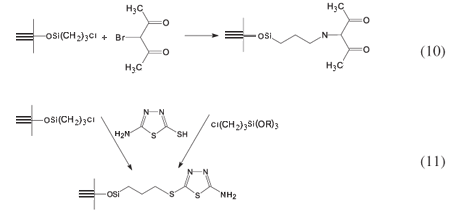
A reatividade do grupo amino pode também ser evidenciada quando da reação com 3-bromopentanodiona para imobilizar o agente quelante b-dicetoamina22, como ilustrado na Equação 10, na reação direta com o complexo de cobre com acetilacetona23 ou através da reação de imobilização da molécula 2-mercaptobenzotiazol, em presença de formaldeído24. Fato marcante é a reação de imobilização do herbicida picloram que, além de diminuir a toxidez da microbiota do solo, age também no decréscimo da biodegradação, porém, tem ainda a capacidade de complexar cátions25-28. Este mesmo comportamento foi observado pelo herbicida derivado do ácido 2,4-diclorofenoxidoacético29. Um aspecto interessante a respeito da distinção de centros potencialmente ativos para interagir com o cloro da matriz foi efetuado com a molécula 5-amino-1,3,4-tiazoltiol, mostrando que a imobilização ocorre pelo grupo tiol do anel tanto através da rota homogênea como heterogênea30, como mostra a Equação 11. Anéis de pirazol imobilizados dão alta versatilidade na remoção de metais pesados com afinidade para o mercúrio31, porém, quando o macrociclo é imobilizado, acontece o ajuste de coordenação do metal no interior do anel32.
Um interessante reagente capaz de provocar o aumento de cadeia é a etilenimina, (H2C)2NH, que apresenta um anel de três membros e, portanto, fácil de ser aberto em presença de um centro básico, como acontece com os agentes sililantes contendo tanto o grupo amino como o tiol. Nestas reações em meios homogêneo ou heterogêneo, partindo do agente com as funções X (NH2 ou SH) chega-se a uma cadeia alifática dispondo de um aumento de centros básicos33-35, com mostra a Equação 12.
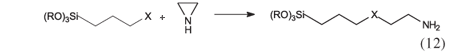
A molécula etilenimina torna-se altamente reativa quando está em presença de grupos silanóis da própria sílica gel. Mesmo estando atento para um critério estequiométrico de grupos silanóis, com uma população da ordem de 5,0 mmol por grama de sílica, os resultados são desencontrados, mas, com excesso do reagente forma-se uma condensação de moléculas sobre a superfície33, como mostra a Equação 13.

Um agente sililante de extrema importância na incorporação de moléculas para dar maior capacitação em agrupar centros básicos é o 3-glicidoxidoalquilsiloxano, que apresenta um anel epóxido de três membros e como acontece com a etilenimina, facilmente é aberto, como na reação com amônia gasosa, etilenodiamina, propile-nodiamina, etanolamina e dietanolamina, como pode ser visto para as moléculas representadas por XNH2 (X=H, (CH2)2NH2, (CH2)3NH2, (CH2)2OH) e NH{(CH2)2OH]2 em um mecanismo proposto36, que leva à incorporação da molécula de interesse, como mostra a Equação 14. Este reagente propicia a obtenção do novo regente sililante, cujo processo de imobilização ser efetuado tanto pelas rotas homogênea como heterogênea10,37,38.

Com o conhecimento progressivo da reatividade da sílica gel foram desenvolvidos paralelamente os princípios para que se pudesse sintetizar a sílica mesoporosa, que devido as suas características se apresenta mais organizada. O sucesso enorme desse material provém da inicial participação da formação de micela, após a mistura de aminas alifáticas em meio aquoso. A hidrólise lenta do tetraetoxidossilano conduz à rede inorgânica depositada sobre a micela, cujo produto de reação é decantado em longo tempo e separado por filtração. O sólido seco pode novamente ser suspenso em um solvente para que receba a imobilização do agente sililante desejado. Essa rota é denominada de pós-modificação, ao contrário daquela em que ambos, o agente formador da estrutura inorgânica e o agente sililante, são hidrolisados conjuntamente através da rota de co-condensação39,40.
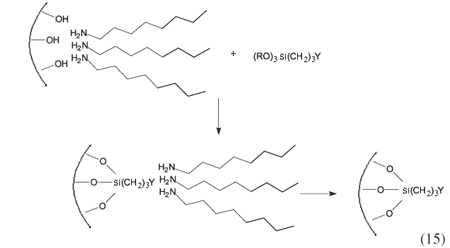
O campo de síntese da sílica mesoporosa cresceu bruscamente nesses últimos anos com introdução de vários compostos na formação de micelas, como surfatantes, copolímeros, compostos sintéticos, ácido láurico, incluindo-se até fios de cabelo humano ou pólen de flores. Porém, o fato marcante provém da remoção do agente micelar direcionador, para deixar livre os espaços internos dos poros ou somente os grupos pendentes originários do agente sililante, na formação dos poros, de acordo com as rotas de sínteses. O caso específico de uma sílica mesoporosa, em que o agente micelar direcionador é uma amina de cadeia alifática longa, n-octilamina, é ilustrado na Equação 15. Acrescenta-se também que a microestrutura da sílica mesoporosa não é bem definida como acontece com os zeólitos e de acordo como as rotas sintéticas têm-se denominações que são MCM-41, SBA-1, SBA-15, MSU, HMS etc, obtidas através do processo de co-condensação. Por outro lado, não só à sílica se aplicam os mesmos princípios de formação do direcionador micelar, mas também a vários outros elementos que possam formar óxidos durante o processo de hidrólise, para que se possa formar a camada inorgânica sobre a micela. Os produtos provenientes desses materiais mesoporosos diferem daquele da sílica e são denominados diferentemente11,40.
Existem várias possibilidades de introduzir moléculas contendo centros de nitrogênio em suas estruturas em sílica mesoporosa MCM-41, obtida pela rota de pós-modificação. Nesse caso tira-se proveito da prévia imobilização do agente sililante clorado com as moléculas de interesse, com uma série de aminas, incluindo poliaminas41,42, como mostra genericamente a Equação 16. Num caso particular, a reação com etilenodiamina (en) leva à confirmação de duas estruturas através de várias técnicas, como ilustra a Equação 17. Levando em consideração a existência de estruturas em pontes entre dois grupos propil, os autores sugerem a formação de quatro possíveis estruturas para a dietilenotriamina (den), Equação 18, e seis outras estruturas são propostas para a trietil-enotriamina (ten), Equação 19. Claro que nestas condições estão consideradas as possibilidades de efeitos interativos exercidos pelos efeitos nucleófilos do nitrogênio básico na interação com o cloro ligado à cadeia do agente imobilizado. A coexistência dessas complexas estruturas parece ser eliminada com a amina terciária 11. Quando o mesmo processo de imobilização foi aplicado na sílica mesoporosa SBA-15 com etilenodiamina, somente uma proposta estrutural foi apresentada e também quando se utilizou a dietilenodiamina três possíveis estruturas foram possíveis de se propor42, como mostra a Equação 20.
Em todos esses atrativos processos de funcionalização para se ter moléculas ancoradas na superfície, a síntese pode ser perfeitamente orientada para se conseguir estruturas sólidas controladas em escala mesoscópica, com grupos seletivos para as reações posteriores, como na remoção de cátions de soluções aquosas e não aquosas, destacando-se, em particular, a importância na remoção cuidadosa do agente direcionador da micela original43-45.
COMPOSTOS LAMELARES
A classe de compostos lamelares vem ocupando uma posição de destaque nos assuntos relacionados à Química Inorgânica, não só do ponto de vista estrutural, como também no estudo de proprie-dades, principalmente no que tange à inserção de moléculas no interior do espaço interlamelar e conseqüentes modificações químicas, além de várias aplicações4. Grande número desses compostos encontrados na natureza é sintetizado com sucesso em laboratório, com algumas vantagens, por ser possível obtê-los com melhores condições de controle e, portanto, sem as esperadas impurezas, até isomórficas2. Um exemplo ilustrativo é relatado com alguns ácidos silícicos, que originalmente encontrados em lagos africanos alcalinos, podem ser sintetizados com alta pureza em laboratório46. Existe uma variabilidade de compostos lamelares que podem ser acrescidos aos ácidos silícicos como os fosfatos inorgânicos, alquil ou arilfosfatos, fosfonatos, arsenatos, sulfatos, crisotila, talco, filossilicatos etc. Do ponto de vista de reatividade, a maioria dessas estruturas apresenta grupos OH expostos na superfície das lamelas, com maior ou menor condição de acessibilidade do reagente em processos reacionais. Esses grupos são responsáveis por efeitos interativos, que acontecem desde a ligação de hidrogênio com inserção de moléculas caracterizando a intercalação47 até a possível modificação química através de agentes sililantes48. Ambos os aspectos interativos genéricos podem ser visualizados conjuntamente na Equação 21, com o característico aumento da distância interlamelar, devido à intercalação da molécula polar ou ancoramento do agente sililante.


Dentro da classe dos silicatos naturais, o filossilicato destaca-se em especial pelos aspectos estruturais, com camadas tetraédricas de composição T2O5 (T = Si4+, Al3+, Fe3+), unidas através do compar-tilhamento com três átomos de oxigênio para formar uma folha ou camada. A quarta posição é ligada à camada adjacente, que tem participação de grupos axiais OH não compartilhados, contendo cátions em vizinhança octaédrica, sendo mais comum o magnésio, alumínio e ferro. No caso em que uma camada tetraédrica se liga a uma octaédrica2,49,50 obtém-se o filossilicato do tipo 1:1 e quando a camada octaédrica está entre as tetraédricas formam o composto do tipo 2:1. Desta forma, a existência de grupos OH facilita a reatividade com os agentes sililantes48,51, identicamente como ocorre com a sílica gel.
A crisotila, amianto ou asbesto é um filossilicato natural fibroso do grupo das serpentinas em estrutura do tipo 1:1, de fórmula empírica Mg3Si2O5(OH)4 , com uma camada de sílica tetraédrica (tridimita) recoberta por camada de hidróxido de magnésio (brucita), com distância interlamelar52 de 730 pm. Como mencionado, a disposição de grupos OH na superfície facilita a imobilização de agentes sililantes no processo de imobilização de moléculas com centros básicos nitrogenados, com finalidade para remover cátions53,54. Por outro lado, quando a sílica é extraída da crisotila, um teor maior de agente sililante é imobilizado, o que possibilita o emprego na remoção de maneira mais efetiva55.
Como acontece na crisotila, também o talco reage com os derivados alcoxissilanos (H3CO)3SiR [R = (CH2)3NH2, (CH2)3NH(CH2 )3NH2 e (CH2)3NH(CH2) 3NH(CH2)3NH2 . Com o primeiro material natural as cadeias pendentes reagem com aldeídos contendo outros centros básicos que, potencialmente, podem ampliar a capacidade em adsorver cátions56. Já com o talco a imobilização ocorre somente na superfície das lamelas, sem aumentar o espaçamento basal57.
No progresso do desenvolvimento na busca de novos materiais sintéticos, o processo sol-gel teve participação efetiva na obtenção à temperatura ambiente de híbridos cristalinos inorgânico-orgânicos58, através da copolimerização do agente sililante de interesse com um alcoxidossilano do tipo M(OR)n, dando enorme versatilidade aos compostos auto-arranjados, situação em que ocorre a inserção de moléculas orgânicas na rede polimérica inorgânica, em escala nanométrica, cujo controle experimental conduz a híbridos com controle de estruturas e porosidades, com formação de materiais meso ou microporosos, composição homogênea, pureza e habilidade na preparação de materiais modulados, como partículas esféricas, fibras ou filmes finos.
Nessas estruturas formadas os átomos de silício provenientes do alcóxido estão inseridos nas estruturas inorgânicas, para formar as redes tetraédricas SiO4, provenientes do agente sililante, para formar a ligação covalente Si-C. O outro componente metálico da rede inorgânica é um cátion divalente, que se acomoda em arranjo em camada, ocupando uma posição octaédrica para formar as famílias59-64 dos tipos 1:1 e 2:1. Esses compostos sintéticos semelhantes às argilas naturais, como esmectita ou mica, têm vantagem por terem composição de alta pureza, além de proporcionarem um controle quantitativo de defeitos estruturais no produto final65. Esses materiais em camadas inorgânico-orgânico são fundamentalmente baseados nos silicatos de magnésio, com estrutura similar à do talco Si8Mg6O20(OH) 4, sendo que o composto final possui a ligação covalente formada, com a função orgânica proveniente do agente envolvido no processo66-69, cujo processo sintético pode envolver agentes sililantes específico contendo cadeias curtas70 ou longas71.
A maioria dessa classe de compostos lamelares organossilicatos possui o magnésio ocupando os sítios octaédricos, seguido pelo níquel66,69-78, com estruturas cristalinas lamelares 2:1, porém outros filossilicatos organofuncionalizados contêm alumínio69,79,80, cobre76, zinco81 e cálcio82 no interior da estrutura inorgânica.
Os filossilicatos sintetizados dos cátions magnésio, níquel, cobre e zinco contendo na estrutura lamelar nas cadeias pendentes as funções amino, tiol ou macrociclos são matrizes úteis para remoção de cátions de solução aquosa na interface sólido/líquido68,70,75-77,81 . O primeiro exemplo envolvendo a reação do agente sililante derivado do glicidóxido para formar o filossilicato de níquel com um macrociclo foi usado para as constantes de equilíbrio para alguns metais da primeira série de transição68 e foram determinados com ênfase nos dados termodinâmicos da interação cátion/centro básico nitrogenados, via calorimetria58,70,77.
Como é de se esperar, a formação de complexos dentro da cavidade lamelar causa um aumento da distância interlamelar e em alguns casos, um tratamento com ácido após a formação do composto de coordenação facilita a recuperação do material de partida, sem quaisquer danos às propriedades de adsorção77 e, dessa maneira, essa classe de compostos cristalinos, de fácil obtenção em condições amenas, desponta como uma promissora fonte no emprego em extração de metais do meio ambiente. Uma outra aplicação dessa classe de filossilicato está associada à catálise de oxidação de alcanos e alquenos, quando um derivado da porfirina é adicionado covalentemente ao suporte inorgânico previamente imobilizado com o grupo amino83.
Os fosfatos inorgânicos4 e os ácidos silícicos84, sendo esse último com fórmula geral Na2O.(4-22)SiO2(5-10)H2 O, apresentam-se com estruturas cristalinas, denominadas makatita, kenamita, magadeíta, keniaíta e octossilicato (ilirita), além de possibilitarem a intercalação de moléculas polares47,85 no interior das cavidades lamelares, também reagiram com os mesmos agentes sililantes nitrogenados, conduzindo à formação de compostos com cadeias orgânicas pendentes entre as estruturas lamelares46,48.
BIOPOLÍMEROS
Na tendência corriqueira de busca de novos materiais, a fonte natural é, sem dúvida, a mais explorada e torna-se mais atrativa quando o processo de isolamento se torna menos dispendioso, principalmente quando é usado tal qual encontrado ou são necessárias poucas operações de tratamento do mesmo, muito embora certas modificações químicas ampliem a utilidade das suas superfícies. Assim, vários materiais podem ter condições de usos1, como na exploração de casca de Ponkan (Citrus reticulata), que é utilizada como suporte para efetivar tanto a troca iônica como precon-centração86,87. Porém, o fascinante biopolímero celulose88, bem como a quitina89, torna-se mais atrativo após as modificações químicas, com introdução de cadeias contendo grupos básicos, para que exerça ações em remoção de cátions90,91 e corante92.
Nos últimos anos, os biopolímeros que têm despertado maior interesse têm sido a celulose88, que provém de várias fontes, e a quitina3, que é proveniente de crustáceos, moluscos, insetos, cogumelos e outros organismos. A quitina é conhecida como o segundo mais abundante biopolímero depois da celulose; é um poli-b(1 ® 4)2-acetamido-(2-desoxi-D-glucopiranose) e quando sofre o processo de desacetilação conduz à quitosana, poli-b(1 ® 4)-2-amino-(2-desoxi-D-gluco-piranose), que do ponto de vista reacional é muito mais atrativa por conter um grupo amino, que propicia a modificação química da estrutura polimérica original. No esqueleto do biopolímero, a quitina apresenta o grupo acetilado (NHCOCH3) e a celulose o grupo OH, na posição 2 do mesmo esqueleto estrutural do anel. Assim, tanto a celulose93 como a quitina94-97 tornam-se muito mais atrativas quando são quimicamente modificadas. No caso de desacetilação da quitina, normalmente em meio básico produz a quitosana, com o grupo amino livre, como mostra a Equação 22, que pode reagir com várias moléculas com a finalidade de tornar o biopolímero com maior disponibilidade de grupos pendentes, para exercer outras atividades e são ainda amplamente empregadas tanto nas formas de pó, esferas ou filmes. As características da celulose são menos atrativas do ponto de vista reacional, porém, é sensível à cloração, que acontece no carbono 6, através do cloreto de tionila, que o torna fonte importante na reatividade subseqüente, como, por exemplo, na reação com etilenodiamina90,91, como mostrado na Equação 23.

Um fato marcante é que a quitina natural representa a mais abundante fonte de nitrogênio encontrada nos oceanos e o segundo material com conteúdo de nitrogênio encontrado no planeta. A extração não causa problemas ao ecossistema, ainda tem todas as vantagens dos polissacarídeos, sendo biopolímero compatível com tecidos animais e baixa toxidez3,89.
Em termos de reatividade a quitosana é muito mais explorada e várias são as reações que, normalmente, acontecem em ácido a-cético diluído, em meio não aquoso ou ainda, em solventes apolares. Normalmente a reação acontece no grupo amino, porém, em condições especiais também pode envolver o carbono 6, como são mostrados na Equação 24, os possíveis produtos de reações89.

INTERAÇÕES METAL/CENTROS BÁSICOS
A insolubilidade tanto dos polímeros inorgânicos como dos biopolímeros em meio aquoso sugere o aproveitamento desses materiais em condições de uso na interface sólido/líquido, numa simulação de aproveitamento em remoção dos metais em um leito reservado e como se sabe, as fases extratoras sólidas são superiores àquelas do sistema líquido-líquido devido à simplicidade, rapidez e habilidade em ser enriquecido pelo metal. Uma vez saturado o extrator, um tratamento adequado de preconcentração do adsorvente precisa ser executado, mas, normalmente, tanto adsorção como desorção são processos cineticamente favoráveis devido à boa porosidade e hidrofilicidade do suporte. O baixo inchamento, boa resistência química e relativamente fácil maneira de ser encontrado esse tipo de material biodegradável na natureza, sendo que alguns casos também favorecem a purificação em tempo curto, são atributos que pesquisadores buscam para isolar e utilizar esses extratores com capacidade de quelar metais de soluções2.
Na adsorção de metais por centros básicos existentes nas cadeias pendentes das superfícies, a saturação pode ser determinada quantitativamente através do processo de batelada, que permite o cálculo do número de moles de cátions em função da concentração do sobrenadante, uma vez previamente determinada a cinética de tempo para se saber sobre o equilíbrio alcançado. O complemento energético dessa interação é obtido através da titulação calorimétrica da mesma reação efetivada na batelada, dentro do recipiente calorimétrico, sempre considerando que as superfícies (SP) em estudo reagem com aos cátions (Mn+)13,46,48, segundo a Equação 25.

Na maioria das vezes essas interações obedecem o modelo de monocamada de adsorção de Langmuir, situação que possibilita a obtenção dos valores de adsorção máxima, variação de entalpia e os correspondentes valores das constantes de equilíbrio. Este último dado propicia o cálculo do valor de energia livre de Gibbs e, conseqüentemente, a entropia do sistema48. Normalmente, os valores de DG são negativos e, portanto, expressam o comportamento espontâneo das reações. Os valores entálpicos associados aos processos de adsorção são normalmente mais favorecidos em solventes não aquosos, sendo quase sempre mais exotérmicos em etanol que em acetona. Também, devido à dessolvatação tanto dos cátions como da própria superfície ancorada, para formar o complexo durante o processo interativo, os sistemas passam a ser favorecidos entropicamente, com a obtenção dos valores positivos. Este conjunto de dados indica que o sistema é termodinamicamente favorável48.
Dentre os agentes sililantes mais comuns empregados na imobilização em sílica gel, para efetuar a remoção de cátions de solução diluída em um determinado solvente, sem dúvida, estão aqueles que contêm grupos nitrogenados2. Em particular, chama atenção o agente contendo a molécula de etilenodiamina, principalmente por exercer propriedade quelante frente aos cátions. No caso dessa molécula contida no agente sililante original ou incorporada num agente sililante precursor13, após a imobilização covalente no interior de lamelas inorgânicas ou em sílica mesoporosa, deixa os grupos pendentes entre as mesmas, o que potencialmente facilita a coordenação. Com a sílica gel, os efeitos interativos em meio etanólico ou acetônico, com alguns metais divalentes da primeira série de transição demonstram que as constantes de equilíbrio são favoráveis, com dois grupos pendentes saturando a coordenação do metal, o que leva aos valores negativos de energia livre de Gibbs, cuja complexação ocorre de maneira espontânea do ponto de vista termodinâmico14. Esse mesmo comportamento de interação ácido/base, tendo genericamente centros básicos nitro-genados pode ser proposto em relação à coordenação sobre a superfície de sílica gel, no interior da cavidade interlamelar de um filosssilicato51, de um composto cristalino lamelar48, em uma cavidade lamelar de um ácido silício46 ou mesmo numa sílica mesoporosa13. Uma representação genérica dos três tipos de superfícies funcio-nalizadas contendo centro amino nas cadeias pendentes de sílica gel, compostos lamelares ou em superfície mesoporosa estão representados nessa seqüência, quando um cátion divalente forma o complexo, conforme a representação na Equação 26.

Um fato marcante acontece com o talco sintético organo-funcionalizado com agentes sililantes nitrogenados de cadeia alifática contendo de um a três átomos de nitrogênio. Os efeitos de remoção de cobre mostraram uma dependência com o número de centros básicos existentes nas cadeias pendentes, ligadas covalentemente ao polímero inorgânico e dependiam ainda do contra ânion usado. A seqüência confirmou que o aumento do número de átomos básicos de nitrogênio causou um significativo aumento na adsorção e o conseqüente aumento da capacidade em formar complexo na superfície modificada, apresentando a seguinte ordem Cl- > NO3- > SO42-. Esta seqüência está relacionada com o raio iônico do cloreto 181 pm, e os raios termodinâmicos do nitrato e sulfato 179 e 258 pm, respectivamente. Esses contra ânions influenciaram no efeito estérico de participação em contrabalancear a carga do complexo formado na superfície, cujo comportamento é muito claro quando se consideram os dados calorimétricos. Os valores entálpicos exotérmicos para os processos interativos seguem a ordem de adsorção e crescem com o número de centros nitrogenados existentes nas cadeias orgânicas. O comportamento não linear da entalpia em função do número de átomos de nitrogênio na molécula, com grande distorção para o ligante tridentado, ilustra o efeito estérico quando se forma o complexo para saturar a coordenação do metal98.
De modo geral, todas essas superfícies modificadas quimicamente são empregadas para a remoção de cátions, normalmente dispersos em um leito aquoso, muito embora algumas aplicações sejam empregadas na remoção de cátions encontrados em álcool comercial, com a finalidade de removê-los, para que causem menor efeito de corrosão, durante o processo de combustão em motores48. Por outro lado, esses efeitos interativos podem ser quantitativamente determinados via calorimetria, com a finalidade de determinações das grandezas termodinâmicas existentes na interface sólido/líquido, sendo que um resumo desses dados está listado na Tabela 1.
Uma expressiva aplicação da quitosana consiste na habilidade em extrair cátions de solução aquosa, através dos efeitos interativos com os grupos amino livres, daí o seu potencial uso em remover cátions99 de um ecossistema, sendo mais comumente utilizado o cobre100, que também teve os efeitos interativos determinados quantitativamente, com ajuste dos dados à tetracoordenação do metal, conforme mostra a Equação 27.

A facilidade de reação do grupo amino livre da quitosana é evidenciada quando na presença de um dialdeído, como é comumente observado com o glutaraldeído, o que facilita a pronta reação para formar ligações cruzadas entre cadeias poliméricas, com a formação da base de Schiff, para dar uma consistência maior ao material101, fato também que pode observado na presença de um polifosfato102. No primeiro caso, a dupla ligação covalente formada pode numa nova etapa ser reduzida, normalmente com boroidreto, para finalizar numa ligação carbono-nitrogênio altamente estável. Na interação com o polifosfato, os efeitos interativos com ambas as extremidades do íon em ponte têm características eletrostáticas e, portanto, sensíveis à mudança de pH. Outro aspecto relevante é a facilidade da quitosana cruzada com glutaraldeído em reagir com enzimas, as quais uma vez imobilizadas podem desempenhar variadas reações catalíticas103,104.
Estudos de remoção de metais consideram vários aspectos do biopolímero: com o zinco105 foi efetuada levando em conta as variáveis de tamanho de partícula, tempo de contato, pH e dose do metal, assim também como um conjunto de metais pesados106, cobre interagindo com oligômeros95 ou com esferas96 e mercúrio com membrana107. No entanto, torna-se muito mais atrativo o uso de quitosana quando acontece a modificação química no grupo amino, condições em que podem ser adicionadas outras moléculas, as quais trazem grupos básicos, para dispor de cadeias pendentes com maior potencialidade em remover cátions de soluções. Desta forma, torna-se atrativa a quitosana modificada com acetilacetona108, hidroxi-benzil109, ácido oxo-glutárico110, oxina111,112, derivados do ácido tetraacético113, piridilmetilideno114 e derivado de formil-fenol115.
Como se nota, várias são as aplicações que podem ser incrementadas com a quitosana, que tem sua aplicação tanto em forma de pó, como esferas ou filmes, dependendo das especificações desejadas. Além do emprego da própria quitosana na remoção de cátions, também pode ser utilizada para remover outros derivativos tóxicos3,116, porém, quando modificada quimicamente a atuação na remoção de cátions de soluções aquosas torna-se mais efetiva, tendo também utilidade em diversas áreas como agricultura, indústria de alimentos, farmacêutica e de cosméticos. As propriedades biológicas da quitosana favorecem o emprego como coagulante, analgésico, de cicatrização, no tratamento de osteo-artrite, efeito hipocolesterolêmico e hipolipidêmico, além da sua discutida ação na remoção de gordura117. Quando associada aos polímeros inorgânicos hidroxiapatita tem a capacidade de uso como reparador de ossos, com sílica em polieletrólitos, porosidade e controle de morfologia; quando intercalada em montmorilonita tem uso na confecção de sensores eletroquímicos ou associada à sepiolita na formação de membranas híbridas118. Uma das aplicações de grande interesse nesses últimos anos está associada à remoção de corantes de um leito líquido, sendo mais empregada a quitosana cruzada119 e com aplicações biomédicas120 ou em forma de esferas121, em especial o azul índigo teve determinada termodinami-camente a interação com a quitosana em pó122.
Os anidridos de ácidos orgânicos têm a capacidade em interagir com a quitosana97, tanto nos carbonos 2 como em 6, assim como com a celulose123 de preferência no carbono 6. Com o anidrido succínico ambas as posições da quitosana sofrem reações, cujo produto final remove cobre de soluções aquosas e possibilita a construção de eletrodo, como empregado em investigações voltamétricas cíclicas. O eletrodo tem grande estabilidade, por mais de um mês sem sofrer quaisquer danos, o que sugere o seu uso como biosensor97,104,124. A quitosana modificada com esse anidrido também tem a capacidade de remover azul de metileno, cujo efeito interativo foi termodinamicamente determinado, recentemente92. Outros aspectos que elucidam a reatividade da quitosana podem ser constatados quando cruzada com glutaraldeído, o que leva a dispor de grupos aldeídicos livres, que podem interagir com agentes sililantes contendo de um a três átomos de nitrogênio na molécula125. No entanto, quando o vidro tem a sua superfície coberta por agentes sililantes, os mesmos interagem com glutaraldeído com subseqüente imobilização da quitosana. O interessante é que esse novo material está associado à capacidade em remover cátions de solução aquosa94.
CONCLUSÃO
Como pôde ser observado no decorrer desta apresentação, várias são as facetas a serem exploradas tanto com os polímeros inorgânicos como com os biopolímeros. Além do enfoque relacionado aos aspectos estruturais, o aproveitamento dos materiais, principalmente no que tange à energética das interações que ocorrem na interface sólido/líquido, cujos valores se prestam muito bem para avaliar a capacidade que têm os polímeros em remover metais pesados de um ecossistema. Dessa forma, quaisquer contribuições neste sentido, com certeza, enriquecerão este fantástico campo que se encontra totalmente convidativo a toda sorte de pesquisa.
Como se trata de sistemas até certo ponto pouco explorados até o momento, o que se busca é atingir as condições preparativas para alcançar o melhor conteúdo propício de centros básicos contidos nas cadeias pendentes dos polímeros. A idealização do produto final aliado aos esforços experimentais pode conduzir adequadamente ao material proposto, direcionado ao uso extrativo dos metais poluentes que afetam o sistema evitando, assim, a contaminação do ecossistema, cuja aplicação leva ao impacto ambiental, de saúde e a programas públicos.
Finalmente, uma tendência que vem se acentuando nesses últimos anos é a possibilidade de agregar moléculas nas superfícies, as quais podem gerar uma seqüência orientada na formação de dendrímeros. Alguns exemplos ilustram essa arquitetura em superfícies, como em sílica gel127 ou mesmo em quitosana128, de rara beleza estrutural e que, por sinal, poderia ser utilizada em remoção de cátions de soluções aquosas ou não aquosas. Como ilustração, quando os filtros cerâmicos são impregnados com dendrímeros adequados, a eliminação de compostos policíclicos aromáticos da água pode ser alcançada através de filtragem129. Como se trata de um campo de pesquisa totalmente aberto, espera-se que despertará maior interesse da comunidade em futuras explorações. Ressalta-se ainda que as determinações energéticas das interações ácido/base, na interface sólido líquido, despontam como um aspecto de relevância nas possíveis separações envolvendo mistura de cátions126,127.
AGRADECIMENTOS
À FAPESP pelo apoio financeiro e ao CNPq pela bolsa. Os alunos e ex-alunos são também agradecidos, em especial F. J. V. A. Oliveira pelo suporte nas elaborações das equações químicas.
Recebido em 20/10/06; aceito em 18/4/07; publicado na web em 19/12/07
- 1. Babel, S.; Kurniawan, T. A.; J. Hazard. Mater 2003, B97, 219.
- 2. Airoldi, C. Em Química de Coordenação Fundamentos e Atualidades; Farias, R. F., ed.; Editora Átomo: Campinas, 2005, cap. 5.
- 3. Crini, C.; Prog. Polym. Sci 2005, 30, 38.
- 4. Alberti, G.; Bein, T.; Comprehensive Supramolecular Chemistry, Pergamon: New York, 1996, vol. 7.
- 5. Airoldi, C.; Santos Junior, L. S.; Thermochim. Acta 1986, 104, 111.
- 6. Duc, M.; Gaboriand, F.; Thomas, F.; J. Colloid Interface Sci 2005, 289, 139.
- 7. Jal, P. K.; Mishra, B. K.; Talanta 2004, 62, 1005.
- 8. Voronkov, M. G.; Vlasova, N. N.; Pozhidaev, Y. N.; Appl. Organomet. Chem 2000, 14, 287.
- 9. Arakaki, L. N. H.; Airoldi, C.; Quim. Nova 1999, 22, 246.
- 10. Sales, J. A. A.; Airoldi, C.; Thermochim. Acta 2005, 427, 77.
- 11. Yoshitake, H.; New J. Chem 2005, 29, 1107.
- 12. Fryxell, G. E.; Wu, H.; Lin, Y.; Shaw, W. J.; Birnbaum, J. C.; Linehan, J. C.; Nie, Z.; Kemner, K.; Kelly, S.; J. Mater. Chem 2004, 14, 3356.
- 13. Sales, J. A. A.; Prado, A. G. S.; Airoldi, C.; Surf. Sci 2005, 590, 51.
- 14. Airoldi, C.; Gushikem, Y.; Espinola, J. G. P.; Colloids Surf 1986, 17, 317.
- 15. Cestari, A. R.; Airoldi, C.; Langmuir 1997, 13, 2681.
- 16. Cestari, A. R.; Vieira, E. F. S.; Simoni, J. A.; Thermochim. Acta 2000, 348, 25.
- 17. Kramer, J.; Driessen, W. L.; Koch, K. R.; Reedijk, J.; Hydrometallurgy 2002, 64, 59.
- 18. Sales, J. A. A.; Faria, F. P.; Prado, A. G. S.; Airoldi, C.; Polyhedron 2004, 23, 719.
- 19. Soliman, E. M.; Mohamoud, M. E.; Ahmed, S. A.; Talanta 2001, 54, 243.
- 20. Mohamoud, M. E.; Soliman, E. M.; Talanta 1997, 44, 15.
- 21. Espínola, J. G. P.; Arakaki, L. N. H.; Oliveira, S. F.; Fonseca, M. G.; Campos Filho, J. A. A.; Airoldi, C.; Colloids Surf 2003, 221, 108.
- 22. Airoldi, C.; Alcântara, E. F. C.; Thermochim. Acta 1995, 259, 95.
- 23. Silva, A. R. ; Wilson, K.; Withwood, A. C.; Clark, J. H.; Freire, C.; Eur. J. Inorg. Chem 2006, 1275.
- 24. Pu, Q.; Su, Z.; Hu, Z.; Chang, X.; Yang, M.; J. Anal. At. Spectrom 1988, 13, 545.
- 25. Prado, A. G. S.; Airoldi, C.; Thermochim. Acta 2002, 394, 155.
- 26. Prado, A. G. S.; Airoldi, C.; Thermochim. Acta 2002, 394, 163.
- 27. Prado, A. G. S.; Airoldi, C.; Anal. Bioanal. Chem 2003, 376, 686.
- 28. Prado, A. G. S.; Tosta, A.; Airoldi, C.; J. Colloid Interface Sci 2004, 269, 259.
- 29. Prado, A. G. S.; Airoldi, C.; Anal. Chim. Acta 2001, 432, 201.
- 30. Prado, A. G. S.; Sales, J. A. A.; Carvalho, R. M.; Rubim, J. C.; Airoldi, C.; J. Non-Cryst. Solids 2004, 333, 61.
- 31. Radi, W.; Ramdani, A.; Lekchiri, Y.; Morcellet, M.; Crini, G.; Janus, L.; Bacquet, M.; New. J. Chem 2003, 27, 1224.
- 32. Dubois, D. ; Tripier, R. ; Bandès, S. Denat, F. ; Guillard, R. ; J. Mater. Chem 2002, 12, 2255.
- 33. Arakaki, L. N. H.; Airoldi, C.; Polyhedron 2000, 19, 367.
- 34. Arakaki, L. N. H.; Espínola, G. P.; Oliveira, S. F.; Freitas, J. M. P.; Gouveia, A. G.; Airoldi, C.; Colloids Surf 2002, 203, 129.
- 35. Airoldi, C.; Arakaki, L. N. H.; J. Colloid Interface Sci 2002, 249, 1.
- 36. Prado, A. G. S.; Arakaki, L. N. H.; Airoldi, C.; Green Chem 2002, 4, 42.
- 37. Sales, J. A. A.; Prado, A. G. S.; Airoldi, C.; Polyhedron 2002, 21, 2647.
- 38. Sales, J. A. A.; Prado, A. G. S.; Airoldi, C.; J. Therm. Anal. Calorim 2002, 70, 135.
- 39. Soler-Illia, G. J. A. A.; Sanchez, C.; Lebeau, B.; Patarin, J.; Chem. Rev 2002, 102, 4093.
- 40. Hoffmann, F.; Cornelius, M.; Morell, J.; Froba, M.; Angew. Chem., Int. Ed 2006, 45, 3216.
- 41. Yoshitake, H.; Koso, E.; Tatsumi, T.; Horie, H.; Yoshimura, H.; Chem. Lett 2004, 33, 872.
- 42. Yokoi, T.; Yoshitake, H.; Tatsumi, T.; J. Mater. Chem 2004, 14, 951.
- 43. Antochshuk, V.; Jaroniec, M.; Chem. Mater 2000, 12, 2496.
- 44. Prado, A. G. S.; Airoldi, C.; J. Mater. Chem 2002, 12, 3823.
- 45. Bourlinos, A. B.; Karakostas, T.; Petridis, D.; J. Phys. Chem. B 2003, 107, 920.
- 46. Ruiz, V. S. O. ; Petrucelli, G. C. ; Airoldi, C. ; J. Mater. Chem 2006, 16, 2338.
- 47. Macedo, T. R.; Airoldi, C.; Microporous Mesoporous Mater 2006, 94, 81.
- 48. Lazarin, A. M.; Airoldi, C.; Chem. Mater 2006, 18, 2226.
- 49. Fonseca, M. G.; Airoldi, C.; Quim. Nova 2003, 26, 699.
- 50. Pomogailo, A. D.; Inorg. Mater. 2005, 41, Suppl. 1 S47.
- 51. Fonseca, M. G.; Barone, J. S.; Airoldi, C.; Clay Clays Miner 2000, 48, 638.
- 52. Wang, L.; Lu, A.; Wang, C.; Zheng, X.; Zhao, D.; Liu, R.; J. Colloid Interface Sci. 2006, 295, 436.
- 53. Fonseca, M. G.; Airoldi, C.; J. Colloid Interface Sci 2001, 240, 229.
- 54. Fonseca, M. G.; Silva Filho, E. C.; Machado Junior, R. S. A.; Arakaki, L. N. H.; Espínola, J. G. P.; Oliveira, F. O.; Airoldi, C.; Colloids Surf, A 2003, 227, 85.
- 55. Fonseca, M. G.; Oliveira, A. S.; Airoldi, C.; J. Colloid Interface Sci 2001, 240, 533.
- 56. Fonseca, M. G.; Airoldi, C.; J. Mater. Chem 1999, 9, 1375.
- 57. Fonseca, M. G.; Airoldi, C.; Mater. Res. Bull 2001, 36, 277.
- 58. Sales, J. A. A.; Petrucelli, G. C.; Oliveira, F. J. V. E.; Airoldi, C.; J. Colloid Interface Sci 2006, 297, 95.
- 59. Plouet, M. R.; Vilminot, S.; Guillot, M.; New J. Chem 2004, 28, 1073.
- 60. Jaber, M.; Brendle, J. M.; Dred, R. L.; J. Mater. Sci 2004, 39, 1489.
- 61. Fujii, K.; Fujita, T.; Iyi, N.; Kodama, H.; Kitamura, K.; J. Mater. Sci. Lett 2003, 22, 1459.
- 62. Fujii, K.; Hayashi, S.; Kodama, H.; Chem. Mater 2003, 15, 1189.
- 63. Guillot, M.; Plouet, M. R.; Vilminot, S.; J. Mater. Chem 2002, 12, 851.
- 64. Plouet, M. R.; Vilminot, S.; Guillot, M.; Kurmoo, M.; Chem. Mater 2002, 14, 3829.
- 65. Fonseca, M. G.; Silva, C. R.; Airoldi, C.; Langmuir 1999, 15, 5048.
- 66. Fukushima, Y.; Tani, M.; J. Chem. Soc., Chem Commun 1995, 241
- 67. Fukushima, Y.; Tani, M.; Bull. Chem. Soc. Jpn. 1996, 69, 3667.
- 68. Hong, Y.; Kim, S.; Bull. Korean Chem. Soc 1997, 18, 236.
- 69. Burkett, S. L.; Press, A.; Mann, S.; Chem. Mater 1997, 9, 1071.
- 70. Fonseca, M. G.; Airoldi, C.; Thermochim. Acta 2000, 359, 1.
- 71. Silva, C. R.; Fonseca, M. G.; Barone, J. S.; Airoldi, C.; Chem. Mater 2002, 14, 175.
- 72. Mizutani, T.; Furushima, Y.; Okada, A.; Kamigaito, O.; Bull. Chem. Soc. Jpn 1990, 63, 2094.
- 73. Whilton, N. T.; Burkett, S. L.; Mann, S.; J. Mater. Chem 1998, 8, 1927.
- 74. Plouet, M. R.; Guillot, M.; Traverse, A.; Chateigner, D.; Vilminot, S.; Nucl. Instrum. Methods Phys. Res., Sect. B 2003, 200, 148.
- 75. Fonseca, M. G.; Silva, C. R.; Barone, J. S.; Airoldi, C.; J. Mater. Chem 2000, 10, 789.
- 76. Fonseca, M. G.; Airoldi, C.; J. Mater. Chem 2000, 10, 1457.
- 77. Fonseca, M. G.; Airoldi, C.; J. Chem. Soc., Dalton Trans 1999, 3687.
- 78. Burattin, P.; Che, M.; Louis, C.; J. Phys. Chem. B 1999, 103, 6171.
- 79. Ukrainczyk, L.; Bellman, R. A.; Anderson, A. B.; J. Phys. Chem. B 1997, 101, 531.
- 80. Jaber, M.; Brendle, J. M.; Delmotte, L.; Dred, R. L.; Microporous Mesoporous Mater 2003, 65, 155.
- 81. Fonseca, M. G.; Silva Filho, E. C.; Machado Jr, R. S. A.; Arakaki, L. N. H.; Espínola, J. G. P.; J. Solid State Chem 2004, 177, 2316.
- 82. Minet, J.; Abramson, S.; Bresson, B.; Sanchez, C.; Montouillout, V.; Lequex, N.; Chem. Mater 2004, 16, 3955.
- 83. Faria, A. L.; Airoldi, C.; Doro, F. G.; Fonseca, M. G.; Assis, M. D.; Appl. Catal., A 2004, 268, 217
- 84. Feng, F.; Balkus Jr, K. J.; J. Porous Mater 2003, 10, 5.
- 85. Machado, M. O.; Lazarin, A. M.; Airoldi, C.; J. Chem. Thermodyn 2006, 38, 130.
- 86. Ajmal, M.; Rao, R. A. K.; Ahmad, R.; Ahmad, J.; J. Hazard. Mater. B 2000, 79, 117.
- 87. Pavan, F. A.; Lima, I. S.; Lima, E. C.; Airoldi C.; Gushikem, Y.; J. Hazard. Mater. B 2006, 173, 527.
- 88. Klemm, D.; Heublein, B.; Fink, H. P.; Bohn, A.; Angew. Chem., Int. Ed 2005, 44, 3358.
- 89. Kurita, K.; Prog. Polym. Sci. 2001, 26, 1921.
- 90. Torres, J. D.; Faria, E. F.; Prado, A. G. S.; J. Hazard. Mater. B 2006, 129, 239.
- 91. Silva Filho, E. C.; Melo, J. C. P.; Airoldi, C.; Carbohydr. Res 2006, 341, 2842.
- 92. Lima, I. S.; Ribeiro, E.S.; Airoldi, C.; Quim. Nova 2006, 29, 501.
- 93. Gurnani, V.; Singh, A. K.; Venkataramani, B.; Talanta, 2003, 61, 889.
- 94. Liu, X. D.; Tokura, S.; Haruki, M.; Nishi, N.; Sakairi, N.; Carbohydr. Polym 2002, 49, 103.
- 95. Rhazi, M.; Desbrières, J.; Tolaimate, A.; Rinaudo, M.; Vottero, P.; Alagui, A.; Polymer 2002 43, 1267.
- 96. Ngah, W. S. W.; Endud, C. S.; Mayanar, R.; React. Funct. Polym 2002, 50, 181.
- 97. Lima, I. S.; Lazarin, A. M..; Airoldi, C.; Carbohydr. Polym 2006, 64, 385.
- 98. Fonseca, M. G.; Airoldi, C.; J. Therm. Anal. Calorim 2001, 64, 273.
- 99. Lima, I. S.; Airoldi, C.; Thermochim. Acta 2004, 421, 133.
- 100. Monteiro Jr, O. A. C.; Airoldi, C.; J. Colloid Interface Sci 1999, 212, 212.
- 101. Monteiro Jr, O. A. C.; Airoldi, C.; Int. J. Biol. Macromol 1999, 26, 119.
- 102. Laus, R.; Laranjeira, M. C. M.; Martins, A. O.; Fávere, V. T.; Pedrosa, R. C. Benassi, J. C.; Geremias, R.; Quim. Nova 2006, 29, 34.
- 103. Airoldi, C.; Monteiro Jr, O. A. C.; Polym. Bull 2003, 50, 61.
- 104. Oliveira, I. R. W. Z.; Vieira, I. C.; Quim. Nova 2006, 29, 932.
- 105. Karthikeyan, G.; Anbalagan, K.; Andal, N. M.; J. Chem. Sci 2004, 116, 119.
- 106. Juang, R. S.; Shao, H. J.; Water Res 2002, 36, 2999.
- 107. Lopes, E. C. N.; Anjos, F. S. C.; Vieira, E. F. S.; Cestari, A. R.; J. Colloid Interface Sci 2003, 263, 542.
- 108. Guillen, M. G.; Sanchez, A. G.; Zamora, M. E.; Carbohydr. Res 1992, 233, 255.
- 109. Baba, Y.; Hirakawa, H.; Yoshizuka, K.; Inoue, K.; Kawano, Y.; Anal. Sci 1994, 10, 601.
- 110. Guibal, E. ; Saucedo, I.; Charrier, M. J.; Delanghe, B.; Le Cloire, P.; Water Res. Technol 1994, 30, 183.
- 111. Inoue, K.; Hirakawa, H.; Ishikawa, Y.; Yamaguchi, T.; Nagata, J.; Ohto, K.; Yoshizuka, K.; Sep. Sci. Technol 1996, 31, 2273.
- 112. Alam S. M.; Inoue, K.; Yoshizuka, K.; Ishibashi, H.; Sep. Sci. Technol 1998, 33, 655.
- 113. Inoue, K.; Yoshizuka, K.; Ohto, K.; Anal. Chim. Acta 1999, 388, 209.
- 114. Hu, D.; Cui, Y.; Dong, X.; Fang. Y.; React. Funct. Polym 2001, 48, 201.
- 115. Justi, K. C.; Laranjeira, M. C. M.; Neves, A.; Mangrich, A. S.; Fávere, V.T.; Polymer 2004, 45, 6285.
- 116. Guibal, E.; Prog. Polym. Sci 2005, 30, 71.
- 117. Silva, H. S. R. C.; Santos, K. S. C. R.; Ferreira, E. I.; Quim. Nova 2006, 29, 776.
- 118. Ruiz-Hitzky, E. Darder, M.; Aranda, P.; J. Mater. Chem 2005, 15, 3650.
- 119. Chiou, M. S.; Li, H. Y.; J. Hazard. Mater. B 2002, 93, 233.
- 120. Berger, J.; Reist, M.; Mayer, J. M.; Felt, O.; Peppas, N. A.; Gurny, R.; Eur. J. Pharm. Biopharm 2004, 57, 19.
- 121. Chiou, M. S.; Ho, P. Y.; Li, H. Y.; Dyes Pigm 2004, 60, 69.
- 122. Anjos, F. S. C.; Vieira, E. F. S.; Cestari, A. R.; J. Colloid Interface Sci 2002, 253, 243.
- 123. Melo, J. C. P.; Airoldi, C.; resultados não publicados.
- 124. Yang, M.; Liu, B.; Shen, G.; Yu, R.; Sens, Actuators, B 2004, 101, 269.
- 125. Airoldi, C.; Monteiro Jr, O. A. C.; J. Appl. Polym. Sci 2000, 77, 797.
- 126. Reynhardt, J. P. K.; Yang, Y.; Sayari, A.; Alper, H.; Adv. Funct. Mater 2005, 15, 1641.
- 127. Grüttner, C.; Böhmer, V.; Casnati, A.; Dozol, J. F.; Reinhoudt, D.N.; Reinoso-Garcia, M. M.; Rudershausen, S.; Teller, J.; Ungaro, R.; Verboom, W.; Wang, P.; J. Magn. Magn. Mater. 2005, 293, 559.
- 128. Kumar, M. N. V. R.; Muzzarelli, R. A. A.; Sashiva, H.; Domb, A. J.; Chem. Rev. 2004, 104, 6017.
- 129. Arkas, M.; Tsiourvas, D.; Paleos, C. M.; Chem. Mater 2005, 17, 3439.
Datas de Publicação
-
Publicação nesta coleção
22 Fev 2008 -
Data do Fascículo
2008
Histórico
-
Recebido
20 Out 2006 -
Aceito
18 Abr 2007

