Resumo
Hydantoin (imidazolidine-2,4-dione) is a 2,4-diketotetrahydroimidazole discovered by Baeyer in 1861. Thiohydantoins and derivatives were prepared, having chemical properties similar to the corresponding carbonyl compounds. Some biological activities (antimicrobial, anticonvulsant, schistosomicidal) are attributed to the chemical reactivity and consequent affinity of hydantoinic rings towards biomacromolecules. Therefore, knowledge about the chemistry of hydantoins has increased enormously. In this review, we present important aspects such as reactivity of hydantoins, acidity of hydantoins, spectroscopy and cristallographic properties, and biological activities of hydantoin and its derivatives.
hydantoin; reactivity; biological properties
hydantoin; reactivity; biological properties
REVISÃO
Estrutura, reatividade e propriedades biológicas de hidantoínas
Structure, reactivity, and biological properties of hidantoines
Silvânia Maria de OliveiraI, - In Memoriam ; João Bosco Paraíso da SilvaI, * * e-mail: paraiso@ufpe.br ; Marcelo Zaldini HernandesII; Maria do Carmo Alves de LimaIII; Suely Lins GaldinoIII; Ivan da Rocha Pitta III
IDepartamento de Química Fundamental, Universidade Federal de Pernambuco, Av. Prof. Luiz Freire, s/n, 50740-540 Recife PE, Brasil
IIDepartamento de Ciências Farmacêuticas, Universidade Federal de Pernambuco, 50740-521 Recife PE, Brasil
IIIDepartamento de Antibióticos, Universidade Federal de Pernambuco, 50670-901 Recife PE, Brasil
ABSTRACT
Hydantoin (imidazolidine-2,4-dione) is a 2,4-diketotetrahydroimidazole discovered by Baeyer in 1861. Thiohydantoins and derivatives were prepared, having chemical properties similar to the corresponding carbonyl compounds. Some biological activities (antimicrobial, anticonvulsant, schistosomicidal) are attributed to the chemical reactivity and consequent affinity of hydantoinic rings towards biomacromolecules. Therefore, knowledge about the chemistry of hydantoins has increased enormously. In this review, we present important aspects such as reactivity of hydantoins, acidity of hydantoins, spectroscopy and cristallographic properties, and biological activities of hydantoin and its derivatives.
Keywords: hydantoin; reactivity; biological properties.
INTRODUÇÃO
Os fármacos são substâncias usadas para impedir ou curar doenças em homens e animais. A introdução de novos fármacos na terapêutica é necessária para o aperfeiçoamento do tratamento de doenças já existentes ou recém-identificadas ou, ainda, para a implementação de tratamentos mais seguros e eficazes. Desde a Antigüidade, diversas civilizações possuíam coleções de produtos naturais que usavam como tratamentos para os diversos males. Em particular, no final do século XIX, a busca por medicamentos menos tóxicos resultou na introdução de substâncias sintéticas na terapêutica e seu uso foi amplamente disseminado no século XX.1 Em estatísticas da área de química medicinal,2 em 2001, cerca de 85% dos fármacos disponíveis na terapêutica moderna são de origem sintética, isto sem considerar aqueles oriundos de processos de semi-síntese. Neste contexto, diversas substâncias sintéticas são obtidas a partir de derivações de anéis heterocíclicos, dentre as quais, destaca-se a hidantoína (Figura 1a) devido à sua potencialidade como protótipo para o desenvolvimento de novos fármacos.
A hidantoína foi descoberta por Baeyer, em 1861, enquanto pesquisava as reações do ácido úrico e corresponde ao 2,4-diceto-tetra-hidro-imidazol, embora seja também denominada imidazolidina-2,4-diona. A primeira fórmula estrutural da hidantoína foi sugerida por Kolbe, em 1870, tendo sido modificada por Strecker, que, neste mesmo ano, propôs uma nova fórmula que é aceita até hoje. A partir dessa época, o sistema anelar hidantoínico tem sido intensivamente estudado tanto no tocante aos aspectos químicos, quanto biológicos.3
A substituição dos átomos de oxigênio carbonílicos da hidantoína por átomos de enxofre origina três tio-derivados: a 2-tio-hidantoína (2-tioxo-imidazolidin-4-ona) (Figura 1b), a 4-tio-hidantoína (4-tioxo-imidazolidin-2-ona) (Figura 1c) e a 2,4-ditio-hidantoína (imidazolidina-2,4-ditiona) (Figura 1d).
A primeira tio-hidantoína sintética, a 2-tio-hidantoína (2-tioxo-imidazolidin-4-ona), foi obtida por Klason (1890) pelo aquecimento do hidrocloreto de etil-aminoacetato em presença de tiocianato de potássio.4 Mais tarde, em 1911, três novos trabalhos publicados por Komatsu, por Wheeler e colaboradores e por Johnson e Nicolet,5 apresentaram novos métodos de preparação desta substância. Johnson e Chernoff6 prepararam a 4-tio-hidantoína, a partir do tratamento da carbetóxi-aminoacetonitrila com H2S em meio básico. Jacobson,7 em 1939, patenteou o método para a síntese de derivados da 2,4-ditio-hidantoína pela reação de uma aminonitrila com o dissulfeto de carbono.
Ware,8 em seu importante artigo de revisão de 1950, relatou que os compostos tiocarbonilados e seus derivados eram mais facilmente sintetizados e isolados que os compostos carbonilados. Por sua vez, a reatividade dos compostos tiocarbonilados era notadamente maior que os derivados com oxigênio. Edward,9 em 1966, em seu artigo de revisão sobre tio-hidantoínas, evidenciou as mudanças produzidas pela substituição do oxigênio por enxofre, principalmente sobre o aumento da polarização do grupo tiocarbonila.
Os derivados hidantoínicos destacam-se por apresentarem ação biológica diversificada, como por exemplo, antimicrobiana, anti-convulsivante e antiparasitária. É digno de nota que a hidantoína tem causado um grande impacto na indústria farmacêutica, visto que vários fármacos contendo este anel heterocíclico são utilizados na clínica. Por exemplo, a 5,5-difenil-hidantoína, denominada genericamente de fenitoína (Dilantin® USA) (Figura 2), é amplamente utilizada no mundo como o anticonvulsivante de escolha no tratamento das crises epilépticas, especificamente de convulsões parciais e tônico-clônicas, mas não nas crises de ausência. Sua atividade antiepilética é, principalmente, associada ao bloqueio dos canais de sódio.10
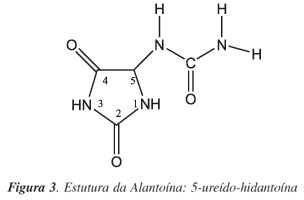
A alantoína ou 5-ureído-hidantoína (Figura 3) possui propriedade cicatrizante, sendo largamente empregada na clínica em vários países, como na Polônia (Alantan®, Septalan®), na Austrália (Egopsoryl®) e nos Estados Unidos (Alphosyl®, Sebical®).
A 1-[(4-nitro-fenil)-5-furfurilideno-amino]-imidazolidina-2,4 -diona (Figura 4) representa um importante miorrelaxante, atuando na redução da liberação de cálcio, comercialmente conhecida como Dantrium®, sendo utilizada em países como Itália, Alemanha, Estados Unidos e Japão.
Fármacos contendo o anel hidantoínico, como os genéricos nifurfolina, nifurtoinol e nitrofurantoína (Figuras 5a, b e c, respectivamente), apresentam propriedades antibacteriana e anti-séptica. A nitrofurantoína, mais utilizada como anti-séptico, devido a sua capacidade de inibir a desidrogenase bacteriana, provocando a inibição da síntese protéica, é amplamente comercializada em diversos países no mundo (Berkfurin®, Furadantin®, Nitrex®).
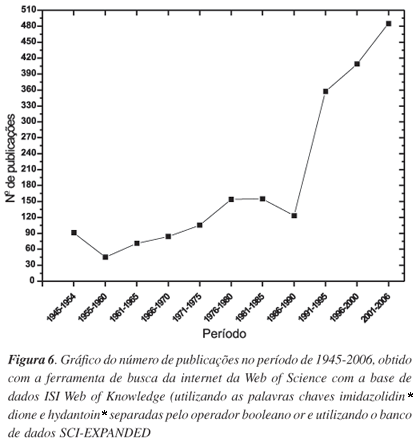
Além das importantes atividades biológicas apresentadas por moléculas derivadas da hidantoína, vários outros aspectos relevantes acerca dessas substâncias têm sido abordados na literatura, cujo foco é dirigido para temas de grande interesse como a reatividade do anel imidazolidínico, a aplicação de novas metodologias de síntese,11 a caracterização estrutural dos compostos, ou ainda, temas relacionados a ensaios biológicos e estudos de relação quantitativa estrutura-atividade QSAR Quantitative Structure Activity Relationships.12 Tais estudos podem ser melhor apreciados graficamente (Figura 6), onde se observa o número de publicações no período de 1945-2006, obtido com a ferramenta de busca da internet da "Web of Science" com a base de dados "ISI Web of Knowledge" (utilizando as palavras chaves "imidazolidin* dione" e "hydantoin*" separadas pelo operador booleano "or" e o banco de dados "SCI-EXPANDED".
REATIVIDADE QUÍMICA DAS HIDANTOÍNAS
De acordo com o artigo de López e Trigo,13 hidantoínas e tio-hidantoínas podem reagir com reagentes eletrofílicos e nucleofílicos. A seguir serão descritos alguns exemplos da reatividade química das hidantoínas.
Protonação
Edward e Liu,14 em 1969, sugeriram que as imidazolidina-2,4-ditionas e as 4-tioxo-imidazolidin-2-onas são primeiramente protonadas nos átomos de enxofre e de oxigênio da posição 2, respectivamente (Figura 7a e 7b). Conclusões similares foram observadas por Congdon e Edward,15 que relataram a formação de cátions hidantoínicos em solução fortemente ácida (Figura 7c e 7d). Em 1980, Edward e Wong16 mostraram igualmente que no caso das 4-tioxo-imidazolidin-2-onas a protonação nos átomos de enxofre e de oxigênio é observada separadamente.
Reações com ácidos de Lewis
De acordo com os trabalhos de Nyitrai e de Markovits-Komis e colaboradores,17 a oxidação e o rearranjo do ácido benzílico ocorrem durante a reação de formação da 5,5-difenil-ditio-hidantoína a partir da benzoína (Figura 8).
As 5,5-difenil-2,4-ditio-hidantoínas, quando submetidas a refluxo com dimetil eterato de trifluoreto de boro ou misturas deste reagente com BF3 em tolueno ou clorobenzeno, são seletivamente metiladas no átomo S2 e/ou sofrem o rearranjo mencionado anteriormente do grupo fenil para fornecer derivados imidazólicos18 (Figura 9).
N-alquilação
As hidantoínas podem ser alquiladas na posição 3 com haletos de alquila tanto em solventes próticos como apróticos. Outros agentes alquilantes incluem dimetil sulfato e diazometano. Hidantoínas mono-alquiladas em N1 podem ser obtidas por proteção do nitrogênio imida com um grupo amino metil, seguido da alquilação do nitrogênio da amida e da remoção do grupo protetor por hidrólise alcalina suave.19 Alquilações em N1 são conhecidas em compostos hidantoínicos substituídos20 na posição 3, contudo, ocorrem em condições mais severas que aquelas em N3. Por outro lado, Trigo e colaboladores,21 em 1978, e Pedregal e colaboradores,21 em 1984, utilizando condições alcalinas ou catálise de transferência de fases, relataram a obtenção de compostos hidantoínicos dissubstituídos nas posições N1 e N3.
Os produtos de alquilação de tio-hidantoínas são cineticamente controlados,22 em relação à possibilidade de tautomerismo nesses compostos S-alquilados. É interessante notar que, dependendo do solvente utilizado, produtos de metilação de 2-tio e 2,4-ditio-hidantoínas existem quase inteiramente em uma das formas tautoméricas23 (Figura 10).
Por outro lado, 2,4-ditio-hidantoínas que apresentam apenas um grupo fenil na posição C5 ocorrem preferencialmente como o tautômero ilustrado na Figura 11, e a sua alquilação ocorre exclusivamente no enxofre da posição 4.14
Hidantoínas contendo grupos metileno ativos
A condensação de hidantoínas que possuem um grupo metileno livre na posição 5 com aldeídos aromáticos foi inicialmente relatada por Wheeler e Hoffman,24 em 1911.
Em 1951, Phillips e Murphy25 relataram quais os principais fatores que poderiam influenciar a reatividade nas reações de condensação: a susceptibilidade do carbono da carbonila a ataques nucleofílicos, relacionada a efeitos eletrônicos e de ressonância; a reatividade do composto que apresenta o grupo metileno, tipo e quantidade de grupos ativadores presentes; a força da base catalítica; as condições da reação, como meio reacional, temperatura e tempo; o grau relativo de estabilização por ressonância dos produtos e, fatores estéricos. Estes autores concluíram que a reatividade do carbono 5 nas reações de condensação com aldeídos aromáticos é diminuída na presença de grupos doadores de elétrons, enquanto grupos retiradores de elétrons produzem um aumento da reatividade, uma vez que o caráter positivo do carbono da carbonila é ampliado.
Baranov e Komaritsa,26 em 1965, através de espectroscopia no ultravioleta, estudaram a cinética de formação de 5-benzilideno-azolidin-4-onas e, caracterizaram o efeito de vários átomos (enxofre, oxigênio e nitrogênio) nas posições 1 ou 2 do anel sobre a reatividade do grupo metileno das azolidin-4-onas. Dentre os resultados encontrados, percebeu-se que a 2-tioxo-imidazolidin-4-ona apresenta uma maior mobilidade do próton metilênico que a imidazolidina-2,4-diona, sendo, portanto, a posição 5 do anel mais reativa para o composto tio-substituído. De fato, em 1980, Moriya, Haigo e Yoneda observaram que as tio-hidantoínas condensam mais rápido que as hidantoínas, sendo as 2-tio-hidantoínas mais reativas que as 2,4-ditiohidantoínas.27
Hidrólise
De acordo com Ware,8 em 1950, hidantoínas e tio-hidantoínas são hidrolisadas em meios alcalinos a ácidos e sob condições vigorosas, formando a-aminoácidos. Este autor cita vários trabalhos sobre o mecanismo de hidrólise dos derivados hidantoínicos. Duas reações paralelas são propostas: o ataque do ânion OH- ao anel hidantoínico livre, ou ao seu ânion no nitrogênio N3.28
Amonólise
As 2-tioxo-imidazolidin-4-onas e as imidazolidina-2-4-dionas reagem com hidrazina e, provavelmente, com aminas, para formar hidrazidas e amidas.29 Por sua vez, derivados da 4-tioxo-imidazolidin-2-ona e imidazolidina-2,4-ditiona reagem com amônia ou aminas na posição 4, fornecendo compostos 4-imino. Algumas imidazolidina-2,4-ditionas reagem com etanolamina na posição 2, formando imino-compostos.30
ACIDEZ DAS HIDANTOÍNAS
As hidantoínas podem ser consideradas ácidos fracos. Para que ocorra a máxima deslocalização da carga do ânion conjugado, a primeira desprotonação deve ocorrer no nitrogênio 3. A 2-tio-hidantoína que apresenta um pKa igual a 8,5 é ligeiramente mais ácida que a hidantoína31 cujo pKa é de 9,12. Entretanto, 2-tio-hidantoínas substituídas na posição 3 podem se ionizar na posição 1 e são ácidos mais fracos (pKa » 11), devido à menor deslocalização da carga do ânion. As 4-tio-hidantoínas são ácidos mais fracos que as 2-tio-hidantoínas.32 Edward e Chin, em 1963, mostraram que as ditio-hidantoínas são ácidos mais fortes que as demais citadas anteriormente, o que foi confirmado por Santos e colaboradores, em 1982, quando encontraram resultados de acidez similares.33
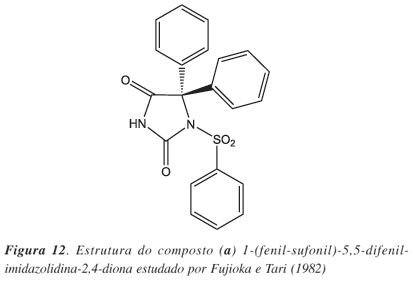
De acordo com Fujioka e Tari,34 em 1982, na maioria dos casos, as constantes de ionização no átomo N3 são pouco afetadas por substituições alquílicas nas posições 1 e 5 do anel imidazolidinônico, embora alguns substituintes no átomo N1 possam influenciar fortemente o pKa. Este é o caso, por exemplo, do composto 1-(fenil-sulfonil)-5,5-difenil-imidazolidina-2,4-diona (1-benzeno-sulfonil-fenitoína) (Figura 12), que possui um pKa de 4,89, enquanto o composto não substituído na posição N1, a 5,5-difenil-imidazolidina-2,4-diona (fenitoína), tem um pKa de 8,31.
Em 1990 e 1991, Bausch e colaboradores35 previram a acidez da imidazolidina-2,4-diona (hidantoína) e 1-metil-imidazolidina-2,4-diona (1-metil-hidantoína) em DMSO, as quais apresentariam valores de pKa = 15,0 e 14,7, respectivamente. Já em água, os valores de pKa destas moléculas seriam 9,0 e 9,1, respectivamente.
Em derivados da imidazolidina-2,4-diona-5,5-dissubstituídos estudados por Kleinpeter e colaboradores,36 os dois prótons NH são ácidos e sofrem uma troca hidrogênio-deutério com adição de água deuterada, em cerca de 2 min. Por outro lado, o estudo RMN 1H de uma solução recém-preparada desses derivados imidazolidínicos em DMSOd6 demonstra que, logo após o primeiro minuto, o próton ligado ao átomo N3 (imida) é completamente trocado, enquanto que o próton ligado ao átomo N1 (amida) continua presente com 35% de intensidade. Estes estudos, somados à observação geral de que o sinal do próton imida N3-H é mais ou menos alargado no espectro de RMN 1H, nos levam a concluir que, nas hidantoínas, o próton ligado ao nitrogênio 3 é mais ácido que aquele ligado ao nitrogênio 1. Comportamento semelhante foi observado por Cristiani e colaboradores37 a partir do estudo das vibrações de estiramento no infravermelho de 5,5-dimetil-imidazolidina-2,4-dionas, em DMSO.
PROPRIEDADES ESPECTROSCÓPICAS
Espectroscopia no ultravioleta
Alguns relatos na literatura se referem aos estudos espectroscópicos na região do domínio ultravioleta de compostos tio-hidantoínicos. Em 1960, Janssen38 estudou absorções de tioamidas no UV, atribuindo a banda de menor intensidade e maior comprimento de onda (Banda I) às transições n®p* do grupo tiocarbonila, que freqüentemente aparece como uma inflexão da banda mais intensa (Banda II), de menores comprimentos de onda, associadas às transições p®p* do grupo tiocarbonila.
A similaridade dos espectros UV de 5-benzilideno-hidantoínas não substituídas e aquelas metiladas nas posições 1 e 3, em solução aquosa, alcoólica e dioxânica, confirma que todas elas existem na forma diceto em solução.39
De acordo com López e Trigo,13 a ionização de 2-tio-hidantoínas em solução alcalina provoca mudanças na absorção. Isto resulta num leve deslocamento hipsocrômico, e um aumento na intensidade da banda principal, além de um deslocamento hipsocrômico e diminuição na intensidade da menor banda.
Espectroscopia no infravermelho
Hidantoínas apresentam duas bandas na região de carbonila em, aproximadamente, 1720 e 1780 cm-1. Entretanto, há uma controvérsia na literatura a respeito da atribuição destas bandas. Alguns trabalhos associam estas bandas aos estiramentos das carbonilas nas posições C2 e C4. Entretanto, a banda de baixa freqüência tem sido atribuída tanto ao grupo carbonila na posição C439,40 quanto à carbonila na posição C2.41 Por outro lado, estudos de espectros de infravermelho e Raman também têm relacionado essas bandas ao acoplamento simétrico e assimétrico entre as vibrações das carbonilas, da mesma forma que ocorre em imidas.42
Comparando o espectro de infravermelho de soluções de 5-benzilideno-hidantoínas e seus derivados N-metil, em solventes de baixa polaridade (dioxano), percebe-se que os sinais de ambas as carbonilas são mantidos, mas são deslocados para freqüências mais altas, indicando que ligações de hidrogênio são quebradas nos derivados N-metil após a dissolução.39
Nyquist e Fiedler,42 em 1995, afirmaram que a hidantoína exibe estas bandas em maior freqüência (1825 e 1785 cm_1) na fase de vapor, enquanto na fase sólida ocorrem em 1783 e 1717 cm_1. Na fase de vapor pode ser observada uma banda em 3495 cm_1, assinalada como um modo de estiramento N-H sem participar de ligação de hidrogênio, já na fase sólida, existem duas bandas em 3150 e 3260 cm_1, assinaladas como modos de estiramento N-H que fazem ligação de hidrogênio.
Kimura e Nagao,43 em 2003, mediram os sinais da vibração de estiramento N-H de derivados da hidantoína em solução de diclorometano. Comparando estas freqüências com aquelas de compostos análogos, como succnimidas, 2-oxazolidonas e hidantoínas substituídas (Figura 13), foram assinalados dois sinais em 3458 e 3426 cm-1, onde o de menor número de ondas foi atribuído ao oscilador N3-H, que é vizinho a duas carbonilas, enquanto o de maior número de ondas, ao N1-H. Este resultado é semelhante ao obtido por Cristiani e colaboradores.37
Os espectros de absorção no infravermelho de 2-tio-hidantoínas apresentam diversas bandas em regiões similares, ocasionando uma grande superposição de bandas e difícil análise do espectro. Uma banda bem definida em 3200 cm-1 é característica das freqüências de estiramento N-H.44 Em 2-tio-hidantoínas ocorre um forte acoplamento entre as vibrações C-S e C-N, resultando em numerosas bandas na região de 1550-1200 cm-1. A identificação na literatura é freqüentemente baseada na banda tiouréia,45 que se situa em 1500 cm-1 quando os substituintes não estão presentes nas posições N1 e N3.
Espectroscopia de ressonância magnética nuclear de 1H e 13C
A espectroscopia de RMN 1H é particularmente utilizada para a localização de substituintes no anel imidazolidínico, considerando os hidrogênios das posições 1, 3 e 5 do anel. Espectros de RMN 1H e de 13C têm sido utilizados com a finalidade de elucidar problemas configuracionais e conformacionais em diversos derivados imidazolidínicos. A seguir são apresentadas algumas destas situações.
Em 1986, Tan e colaboradores46 estudaram o isomerismo geométrico da dupla ligação exocíclica ao anel imidazolidinônico a partir de análises espectroscópicas de 5-benzilideno-imidazolidina-2,4-dionas (Figura 14) e os correspondentes derivados 3-metil e 1-metil substituídos. Estes pesquisadores verificaram, nas reações de condensação com aldeídos que, para os compostos não metilados em N1, apenas um dos isômeros é formado (Z); enquanto que, para os compostos metilados na posição N1 as duas formas isoméricas (Z) e (E) são possíveis. No espectro de RMN de 1H dessas 5-benzilideno-imidazolidina-2,4-dionas, as maiores diferenças referem-se aos deslocamentos químicos dos prótons: vinílicos, ligados ao átomo N1 e orto-fenílicos. O efeito anisotrópico do grupo C4=O desblinda o próton vinílico no isômero Z e blinda os prótons orto-fenílicos quando comparado ao isômero E. O próton ligado a N1 no isômero Z entra em ressonância em campo mais baixo, devido a sua proximidade com o anel aromático, que no isômero E. O sinal em campo mais baixo refere-se ao próton ligado ao átomo N3, devido à influência das duas carbonilas. Como era de se esperar, este próton é pouco afetado pelas diferenças nas configurações Z e E.
Derivados 5-aril-metileno-imidazolidina-2,4-dionas e 5-aril-metileno-2-tioxo-imidazolidin-4-onas (Figura 15) apresentando potencialidade como inibidores de aldose redutase foram estudados espectroscopicamente por Fresneau e colaboradores,47 com a finalidade de determinar a configuração da dupla ligação etilênica exocíclica, por meio de espectroscopia de RMN 1H e 13C. Dentre os 28 compostos estudados, existiam compostos substituídos em N1, N3 e compostos não-substituídos. Verificou-se que a configuração Z é preferencial para os compostos não-substituídos na posição N1 e substituídos na posição N3, enquanto para os compostos com substituintes no átomo N1, a configuração preferencial é a E. Estes resultados foram obtidos baseados no fato de que as estruturas químicas dos compostos N1 substituídos mostram uma potencial interação por efeito homonuclear NOE48Nuclear Overhauser Effect entre o próton etilênico (H6) os prótons metilênicos do fragmento acético (H6-C6=C5-N1-CH2-CO2 H), interação que só poderia ser observada para o isômero E.47
Ösz e colaboradores,49 em 1998, propuseram uma caracterização do tautomerismo de hidantoínas e tio-hidantoínas substituídas nas posições 1 e 5 em solução (DMSOd6) usando constantes de acoplamento em RMN de 13C e 1H. Estes autores concluíram que todos os derivados estudados apresentaram, exclusivamente, a forma tautomérica bis-lactâmica. Ainda observaram que os átomos do anel heterocíclico são coplanares para estes derivados.
Em 1997, Kleinpeter e colaboradores36 afirmaram que a distribuição eletrônica das hidantoínas é importante para a sua atividade biológica e, por esta razão, como parte de um estudo de relações estrutura-atividade para derivados hidantoínicos, o primeiro passo foi a realização de uma análise estrutural detalhada por espectroscopia de RMN de 13C e 1H de uma série de hidantoínas 5,5-dissubstituídas. Adicionalmente, realizaram cálculos teóricos semi-empíricos (AM1 e PM3) e ab initio (RHF/3-21G e RHF/3-21G*, quando o átomo de enxofre está presente) para complementar a análise espectroscópica por RMN, bem como para calcular características estruturais dos derivados estudados. Os dois prótons N1-H e N3-H, nos derivados estudados, absorvem em duas faixas diferentes: 7,5-9,5 ppm e 11,0-11,5 ppm, respectivamente. Adicionalmente, mostraram que esse resultado é concordante com a análise da distribuição de cargas obtidas a partir de cálculos de orbitais moleculares em nível HF/3-21G* para o anel imidazolidinônico não substituído, mostrando que o átomo de hidrogênio ligado a N3 é mais positivo que aquele ligado ao átomo N1, sendo, portanto, o próton ligado a N3 mais ácido e mais desblindado. Através de resultados obtidos pelo experimento Rotating Frame Overhauser Effect Spectroscopy (ROESY)47 realizado para um dos compostos, e do espectro de RMN de 1H acoplado a 15N, concluiu-se que as hidantoínas estudadas existem na forma tautomérica lactâmica. Resultados obtidos por meio de cálculos semi-empíricos AM1 e PM3 para a hidantoína não substituída mostraram que este tautômero (forma lactâmica) é mais estável.36 Com relação às ressonâncias das carbonilas, o assinalamento foi feito por meio de experimentos de RMN 2D otimizados com acoplamentos a longa distância Heteronuclear Multiple Bond Correlation (HMBC),48 onde se observou que os dois carbonos da carbonila absorvem em campo baixo, bem separados em 156-158 ppm (C2=O) e 175-179 ppm (C4=O).
Em 1998, Benassi e colaboradores,50 através de estudos de RMN uni e bidimensionais Correlated Spectroscopy (COSY H, H),48Heteronuclear Multiple Quantum Correlation (HMQC), Heteronuclear Multiple Bond Correlation (HMBC)48 e Rotating Frame Overhauser Effect Spectroscopy Bidimensional (ROESY-2D),48 além de cálculos químico-quânticos, estudaram a estrutura de hidantoínas 3-fenil-5-benzilideno-substituídas. As posições N1-H e N3-fenil foram assinaladas e a possibilidade de tautomerismo foi investigada, um NOE distinto entre o próton ligado a N1 e os prótons orto-fenílicos do grupo benzilideno comprovaram a presença da forma tautomérica bis-lactâmica. A configuração Z da dupla ligação exocíclica (C=C), através de um experimento ROESY, foi assinalada inequivocamente para todos os compostos. Por meio de análise comparativa dos resultados obtidos por cálculos teóricos e os resultados experimentais de RMN, foi encontrada uma conformação onde tanto o grupo fenil da posição 3, quanto o grupo benzilideno da posição 5 ficam torcidos em relação ao plano do anel hidantoínico, semelhante aos resultados teóricos AM1 e ab initio HF/3-21G** obtidos para derivados 3-benzil-5-benzilideno-hidantoínicos.51
Benassi e colaboradores,52 em 1999, realizaram um estudo estrutural por RMN 1H e 13C de 5-benzil-imidazolidina-2,4-dionas em solução. Os deslocamentos químicos foram relatados e as constantes de acoplamento vicinais H,H e H,C das imidazolidina-2,4-dionas foram empregadas para o cálculo das populações rotaméricas ao longo da ligação C5-benzil. Considerando também as constantes de acoplamento vicinal dos prótons metilênicos com o carbono C4 (JH,Cgauche =1,8 Hz e JH,Ctrans = 6,5 Hz), foi obtida uma conformação onde o grupo benzil da posição 5 é perpendicular ao plano do anel hidantoínico, semelhante ao resultado obtido para a estrutura do estado sólido.53
Cristalografia de raios-X
Diversos trabalhos referem-se a estudos de difração de Raios-X, com objetivo de esclarecer e confirmar a estrutura de compostos imidazolidínicos e seus derivados. Por exemplo, a possibilidade de existência de duas formas tautoméricas do derivado S-metilado da 5,5-difenil-2-tioxo-imidazolidin-4-ona, que já foram anteriormente destacadas neste trabalho, foi comprovada pela estrutura cristalográfica de raios-X.54
O anel hidantoínico é essencialmente planar e uma grande variedade de ligações de hidrogênio é observada em estruturas cristalográficas. A estrutura sólida da 2-tio-hidantoína apresenta inúmeras folhas paralelas empacotadas, onde cada uma delas é formada por uma rede de unidades deste heterociclo interagindo por meio de ligações de hidrogênio N-H...O, onde cada átomo de oxigênio aceita duas ligações de hidrogênio, sendo cada uma delas com um átomo de hidrogênio ligado a N1 e N3 de moléculas vizinhas no cristal. As distâncias de ligações entre os átomos pesados das ligações de hidrogênio N1-H...O4 e N3-H...O4 na 2-tio-hidantoína assumem valores de 2,928 e 2,916 Å, respectivamente. É interessante destacar que esta estrutura não mostrou ligações do tipo N1-H...S=C.54 Entretanto, outros derivados 2-tio-hidantoínicos apresentam este tipo de ligação.55
Uma análise da estrutura cristalográfica da 5,5-dimetil-imidazolidina-2,4-diona realizada por Cassady e Hawkinson,56 em 1982, mostrou que as moléculas são organizadas por interações de empacotamento e por ligações de hidrogênio do tipo N1-H...O=C2 e N3-H...O=C2. O oxigênio do grupo C4=O aparentemente não participa de ligações de hidrogênio no cristal. Neste caso, também se observa que a ligação com o átomo de hidrogênio ligado ao nitrogênio N3 é mais forte que em N1.
A partir de uma análise da estrutura cristalográfica de derivados 5,5-dimetil-imidazolidina-2,4-ditiona; 5,5-dimetil-2-selenoxo-imidazolidin-4-ona; 5,5-dimetil-4-selenoxo-imidazolidin-2-tiona e 2-tio-hidantoína, Devillanova e colaboradores,57 verificaram que todas as moléculas estudadas são essencialmente planares e que os comprimentos das ligações C=X e C=Y são afetados pela natureza do calcogênio, por exemplo: C=O » 1,20 Å; C=S » 1,65 Å; C=Se » 1,80 Å.
Drew e colaboradores,58 em 1987, assinalaram a configuração Z para o derivado 5-benzilideno-1-metil-imidazolidina-2,4-diona. Para esta molécula, a presença do grupo CH3 em N1 torna impossível a sua planaridade. O anel benzênico é rotacionado em aproximadamente 46º em relação ao anel imidazolidínico, existindo, portanto, duas conformações equivalentes para o isômero Z.
Duas estruturas cristalográficas de compostos benzilideno-imidazolidínicos 3,5-dissubstituídos foram determinadas por difração de raios-X, em ambas as estruturas cristalográficas dos compostos 3-(4-bromo-benzil)-5-(4-flúor-benzilideno)-imidazolidina-2,4-diona 59 e 3-benzil-5-(2-flúor-benzilideno)-imidazolidina-2,4-diona ,60 comprovando-se a presença de ligações de hidrogênio intermoleculares N1-H....O2 em fase condensada. Para a primeira, o anel heterocíclico e o grupo benzilideno encontravam-se coplanares, já para o derivado com o flúor na posição 2, o ângulo diédrico entre o anel imidazolidínico e o grupo benzilideno foi de 160,5º.
Em 2004, Yu e colaboradores,61 a partir de uma pesquisa no Cambridge Structural Database, observaram que as hidantoínas formam duas cadeias unidas por ligações de hidrogênio N-H...O, a partir das quais centros de inversão criam uma cadeia de anéis. Dos 50 grupos hidantoínicos em 49 moléculas independentes pesquisadas (uma das quais apresenta mais que uma estrutura hidantoínica), 5 mostram este rearranjo, enquanto 6 apresentaram ligações de hidrogênio onde o átomo de oxigênio O2 simultaneamente aceita N1-H...O2 e N3-H...O2, enquanto o átomo O4 não aceita nenhuma. Hidantoínas também podem formar cadeias infinitas, com o átomo N1 como doador e O4 como aceptor e N3 como doador e O2 como aceptor, e este tipo de rede apresentou 5 ocorrências na pesquisa citada. Muitos derivados hidantoínicos possuem substituintes na posição 5 que desviam algumas ou todas as ligações de hidrogênio, desta forma, 11 moléculas apresentaram N1 como doador e O2 como aceptor, 7 envolveram N3 como doador e O2 como aceptor, 2 com N3 e O4, e uma com participação heterogênea de N1 com O4 e N3 com O2. Adicionalmente, 12 cadeias simples foram observadas e, por fim, 3 anéis interagem apenas com grupos laterais, sem apresentar ligações de hidrogênio entre anéis hidantoínicos.
PROPRIEDADES BIOLÓGICAS DAS IMIDAZOLIDINAS
Nesta seção serão discutidas algumas das diversas atividades biológicas apresentadas por compostos imidazolidínicos.
Ação antimicrobiana
Compostos contendo o sistema hidantoínico possuem propriedade antifúngica e antibacteriana: a iprodiona [3-(3,5-diclorofenil)-N-isopropil-2,4-dioxo-imidazolidina-1-carboxiamida] constitui o fungicida hidantoínico mais importante, inibindo ao mesmo tempo a germinação de esporos e o crescimento de micélios de fungos.62 Por outro lado, as moléculas 5-(aril-metileno)-hidantoínas e 5-(aril-metileno)-2-tio-hidantoínas, obtidas pela condensação de aldeídos aromáticos com hidantoínas ou tio-hidantoínas, apresentam atividades fungicida e bactericida.63
Góes e colaboradores,64 em 1991, relataram a ação antibacteriana apresentada por derivados 5-benzilideno-3-fenil-imidazolidinônicos frente ao Streptococus faecalis e Mycobacterium smegmatis, tendo sido também observada a ação antifúngica de alguns derivados desta série frente aos fungos Candida albicans e Neurospora crassa.
Em 1992, Lima e colaboradores65 estudaram a atividade antimicrobiana de derivados 3-(4-cloro-benzil)-5-benzilideno-imidazolidinônicos e observaram, através da determinação da Concentração Mínima Inibitória (CMI), que o derivado com o substituinte NO2 em posição 4 no grupo benzilideno foi ativo contra Candida albicans e Neurospora crassa e também frente às bactérias Staphylococus aureus, Escherichia coli e Mycobacterium smegmatis.
Cavalcanti Amorim e colaboradores,66 em 1992, determinaram a atividade antimicrobiana de compostos da série 3-(4-bromo-benzil)-5-benzilideno-imidazolidina-2,4-diona. Os resultados mostraram uma atividade antifúngica variando de 30 a 50 µg/mL para os compostos testados para Candida albicans e Neurospora crassa, enquanto a CMI para o Mycobacterium smegmatis ficou entre 10 e 50 µg/mL.
O composto 3-(4-cloro-benzil)-4-tioxo-5-arilazo-imidazolidin-2-ona foi sintetizado e sua atividade microbiana foi avaliada, mostrando-se ativo frente ao Bacillus subtilis, Bacillus anthracis e Serratia marcenscens, dentre outros microrganismos.67
Em 1999, Albuquerque e colaboradores,68 sintetizaram e avaliaram a atividade antimicrobiana de derivados 3-benzil-5-arilideno-imidazolidinônicos. Dez compostos foram avaliados como potenciais agentes antimicrobianos frente a seis microrganismos (Staphylococus aureus, Micrococus flavus, Bacillus cereus, Proteus vulgaris, Salmonella enteritidis e Escherichia coli) e apresentaram uma CMI entre 32 e 128 µg/mL.
Ação no sistema nervoso
No que se refere à ação no sistema nervoso, são conhecidas também diversas atividades biológicas, tanto nas hidantoínas, como nas tio-hidantoínas. Por exemplo, Henze e Smith,69 em 1943, descobriram que a 2,4-ditio-5,5-dimetil-hidantoína possuía ação hipnótica. Este composto revelou também atividade anticonvulsivante superior à 5,5-dimetil-hidantoína. Outros trabalhos também demonstraram que derivados 2-tioxo-imidazolidina-4,5-dionas possuíam atividade anticonvulsivante.70 Novas hidantoínas substituídas, como 4-hidróxi-2-imidazolidinonas, 2-imidazolonas, 2-imidazolidinonas, diaminas vicinais e derivados de aminoácidos, foram sintetizadas e avaliadas quanto à atividade anticonvulsivante por Cortes e colaboradores71 (1985). Estes estudos demonstraram que a seqüência de átomos N-C-C-N-C presente nas hidantoínas era essencial para esta neuroatividade.
Danielson e Sundell,72 em 1965, verificaram que compostos que continham a estrutura 2-tioxo-imidazolidina-4,5-diona apresentavam atividade sedativa. A partir desses estudos, Ozkirimli e Hamali,73 em 1995, sintetizaram e testaram a atividade hipnótico-sedativa dos derivados 2-tioxo-imidazolidina-4,5-dionas, tendo o composto 1-(benzilideno-amino)-3-ciclo-hexil-2-tioxo-imidazolidina-4,5-diona apresentado maior atividade.
Karali e colaboradores,74 em 1998, obtiveram derivados com atividade anticonvulsivante, onde o composto 1-alil-3-(2-oxo-1,2-diidro-indol-3-ilidenoamino)-2-tioxo-imidazolidina-4,5-diona foi identificado como o mais potente.
A difenil-hidantoína, denominada genericamente de fenitoína, é amplamente utilizada como o anticonvulsivante de escolha no tratamento das crises epilépticas.75 Este fármaco, por sua eficácia já comprovada, faz parte da Relação de Medicamentos Essenciais da Organização Mundial de Saúde76 (WHO) e também da Relação Nacional de Medicamentos Essenciais.77
A influência das características estruturais e da lipofilicidade de derivados hidantoínicos sobre a atividade anticonvulsivante foi investigada por Scholl e colaboradores,78 em 1999. As lipofilicidades de 75 hidantoínas determinadas experimentalmente por RP-HPLC foram correlacionadas com valores estimados teoricamente a partir de estruturas obtidas por modelagem molecular. Foi então proposto um modelo farmacofórico para a interação dos derivados hidantoínicos com o canal de sódio, envolvendo um aromático, um sítio aceptor e um sítio doador de ligação de hidrogênio fazendo parte de um sistema quiral de três pontos.
Em 2004, Zha e colaboradores,79 com o intuito de investigar a importância do anel hidantoínico como bloqueador de canais de sódio, avaliaram as afinidades de ligação ao canal de sódio dependente de voltagem neuronal de compostos não-hidantoínicos, como hidróxi-amidas, oxazolidina-dionas, hidróxiácidos e aminoácidos. Os resultados demonstraram que o anel hidantoínico é relativamente importante para compostos com pequenas cadeias laterais, sendo, portanto, menos relevante para compostos com longas cadeias hidrofóbicas. Um outro aspecto relevante, é que são observadas duas regiões farmacofóricas hidrofóbicas, o que está de acordo com o modelo de Khodorov,80 e uma terceira região hidrofóbica adicional pode aumentar a eficiência da interação e fornecer inibidores mais potentes.
Em 1999, Kanyonyo e colaboradores,81 sintetizaram e avaliaram 24 derivados 3-alquil-5,5-difenil-imidazolidina-2,4-diona como ligantes de receptores canabinóides e concluíram que alguns derivados exibiram afinidade superior ao composto amino-alquil-indol de referência. Adicionalmente, foram evidenciadas as suas propriedades antagonistas ao receptor canabinóide CB1. Posteriormente, derivados 3-substituídos-5,5-difenil da imidazolidina-2,4-diona e da 2-tioxo-imidazolidin-4-ona, já descritos previamente82 como ligantes de receptores canabinóides CB1, foram avaliados por Muccioli e colaboradores,83 em 2006 , como inibidores da enzima FAAH (Fatty Acid Amide Hydrolase); observou-se que os derivados da 2-tioxo-imidazolidin-4-ona são mais potentes que os derivados oxo correspondentes. Estes derivados atuam como inibidores competitivos da FAAH, sem ser hidrolisados pela enzima. Dentre eles, o composto 5,5-difenil-tetradecil-2-tioxo-imidazolidin-4-ona apresentou a mais alta atividade frente à enzima FAAH.
Atividade esquistossomicida
Luttermoser e Bond84 observaram que a 5,5-difenil-hidantoína e a 5-(p-clorofenil)-5-metil-hidantoína apresentavam atividade frente a vermes adultos de Schistosoma mansoni em ratos portadores da esquistossomose mansônica.
A 5-(2,4,5-triclorofenil)-hidantoína e a 1-(5-nitro-tiofeno-3-il)-imidazolidin-2-tiona também possuem atividade esquistossomicida.85
Oliveira e colaboradores86 avaliaram a susceptibilidade in vivo do S. mansoni frente a derivados imidazolidinônicos 3,5-dissubstituídos, tendo o composto 3-(4-cloro-benzil)-5-(4-nitro-benzilideno)-imidazolidina-2,4-diona apresentado uma importante atividade esquistossomicida in vitro.
Em 2005, Albuquerque e colaboradores,87 avaliaram a atividade esquistossomicida de derivado tio-hidantoínicos e a mobilidade dos vermes (S. mansoni) foi reduzida após 24 h de contato com as drogas. Também foi observado que os compostos 3-benzil-5-(4-flúor-benzilideno)-1-metil-2-tioxo-imidazolidin-4-onas demonstraram baixos níveis de toxicidade quando administrados em altas doses nos camundongos.
Devido à sua reatividade química e conseqüente afinidade por biomacromoléculas, ao núcleo hidantoínico são atribuídas diversas atividades biológicas, além daquelas já citadas neste artigo. Recentemente, outras atividades estão sendo relacionadas a este heterociclo (antiinflamatória,88 moduladora de colesterol HDL,89 inibidora de agregação plaquetária,90 antiarrítmica91 e anticâncer92 e antiviral).93
CONCLUSÕES
Como demonstrado nesta revisão, a quantidade de trabalhos sobre o núcleo hidantoínico e seus derivados é notadamente crescente nas últimas cinco décadas. Além de trabalhos em físico-química orgânica, vários estudos de Química Medicinal vêm se destacando na literatura. Em particular, vale destacar a contribuição que pesquisadores brasileiros têm oferecido para a compreensão deste sistema. Por exemplo, já na década de 90, em nosso grupo de pesquisa, Lima e colaboradores,94 mostraram que parâmetros lipofílicos e eletrônicos explicam a variância das atividades biológicas hipoglicemiantes de uma série de hidantoínas e tiazolidinas. Dezessete anos depois, os compostos de referência para o tratamento de diabetes mellitus (Tipo II) continuam sendo baseados nos anéis tiazolidínicos. Além disto, a contribuição para o estudo de doenças negligenciadas como, por exemplo, a esquistossomose, justifica o interesse aprofundado dos pesquisadores nacionais para com estes heterociclícos. Novos trabalhos, relacionando a aplicação de modelagem molecular como ferramenta para estabelecer relações quantitativas entre a estrutura química e a atividade biológica QSAR têm direcionado a atenção deste fármaco para problemas relacionados ao sistema nervoso.95 É, portanto, baseado nesta trajetória evolutiva de trabalhos de pesquisa que se pode concluir que esta área é bastante promissora, visto que ainda há muito a se explorar nas propriedades químicas e biológicas da hidantoína e seus derivados.
REFERÊNCIAS
1. Thomas, G.; Química Medicinal Uma Introdução, 1ª ed., Guanabara Koogan: Rio de Janeiro, 2003.
2. Ellestad, G. A.; J. Med. Chem. 2006, 49, 6627; Barreiro, E. J.; Fraga, C. A. M.; Química Medicinal - As Bases Moleculares da Ação dos Fármacos, 1ª ed., Artmed: Porto Alegre, 2001.
3. Baeyer, A.; Ann. Chem. 1861, 117, 178; Finkbeiner, H.; J. Org. Chem. 1965, 30, 3414.
4. Klason, P.; Chem. Ztg. 1890, 14, 543.
5. Komatsu, S.; Mem. Coll. Sci. Eng. Kyoto Imp. Univ. 1911, 3, 1; Wheeler, H. L.; Nicolet, B. H. ; Johnson, T. B.; Am. Chem. J. 1911, 46, 456; Johnson, T. B.; Nicolet, B. H.; J. Am. Chem. Soc. 1911, 33, 1975.
6. Johnson, T. B.; Chernoff, L. H.; J. Am. Chem. Soc. 1912, 34, 1208.
7. Jacobson, R. A.; U.S. Pat. 2 143, 186 1988.
8. Ware, E.; Chem. Rev. 1950, 46, 403.
9. Edward, J. T.; Chem. Org. Sulfur. Comp. Eng. 1966, 2, 287.
10. Rogawski, M.; Porter, R. J.; Pharmacol. Rev. 1990, 42, 223.
11. Meusel, M.; Gütschow, M.; Org. Prep. Proced. Int. 2004, 36, 391.
12. Gaudio, A. C.; Quim. Nova 1995, 19, 278.
13. López, C. A.; Trigo, G.; Adv. Heterocyclic Chemistry 1985, 38, 177.
14. Edward, J. T.; Liu, J. K.; Can. J. Chem. 1969, 47, 1117.
15. Congdon, W. I.; Edward, J. T.; Can. J. Chem. 1972, 50, 3767.
16. Edward, J. T.; Wong, S.; Can. J. Chem. 1980, 57, 15.
17. Nyitrai, J.; Acta Chim. Acad. Sci. Hung. 1967, 51, 95; Markovits-Komis, R.; Nyitrai, J.; Lempert, K.; Chem. Ber. 1971, 104, 3080.
18. Fetter, J.; Nyitrai, J.; Lempert, K.; Tetrahedron 1971, 27, 5953.
19. Orazi, O. O.; Corral, R. A.; Experientia 1965, 21, 508.
20. Orazi, O. O.; Corral, R. A.; Schttenberg, H.; J. Chem. Soc. Perkin Trans. 2 1974, 219.
21. Trigo, G. G.; Avendaño, C.; Varela, I.; Martinez, M.; An. Quim. 1978, 74, 1090; Pedregal, C.; Trigo, G. G.; Espada, M.; Elguero, J.; J. Heterocycl. Chem. 1984, 21, 477.
22. Carrington, H. C.; Waring, W. S.; J. Chem. Soc. 1950, 354.
23. Edward, J. T.; Liu, J. K.; Can. J. Chem. 1972, 50, 2423.
24. Wheeler, L.; Hoffman, C.; Am. Chem. J. 1911, 45, 368.
25. Phillips, A. P.; Murphy, J. G.; J. Org. Chem. 1951, 16, 954.
26. Baranov, S. N.; Komaritsa, I. D.; Khim. Geterosikl. Soedin.1965, 1, 69.
27. Moriya, T.; Hagio, K.; Yoneda, N.; Chem. Pharm. Bull. 1980, 28, 1891.
28. Vogels, G. D.; Windt, F. E.; Bassi, W.; Recl. Trav. Chim. Pays. Bas. 1969, 88, 940; Ivin, B. A.; Rutkowskii, G. V.; Sochilin, E. G.; Tsereteli, Y. U.; Zhr. Org. Khim. 1972, 8, 840; Ivin, B. A.; Rutkowskii, G. V.; Andreev, S. A.; Sochilin, E. G.; Zhr. Org. Khim. 1973, 9, 2194; Ivin, B. A.; Rutkowskii, G. V.; Rusavskaya, T. N.; Sochilin, E. G.; Zhr. Org. Khim. 1975, 11, 2188; Blagoeva, I. B.; Pojarlieff, I. G.; Dimitrinov, V. S.; J. Chem. Soc. Perkin Trans. 2 1978, 887.
29. Edward, J. T.; Nielsen, S.; J. Chem. Soc. 1957, 5084.
30. Rakowitz, D.; Maccari, R.; Ottanà, R.; Vigorita, M.G.; Bioorg. Med. Chem. 2006, 14, 567.
31. Zief, M.; Edsall, J. T.; J. Am. Chem. Soc. 1937, 59, 2245; Pickety, L. W.; MClean, M.; J. Am. Chem. Soc. 1939, 61, 423.
32. Stuckey, R. E.; J. Chem. Soc. 1947, 331.
33. Edward, J. T.; Chin, O. J.; Can. J. Chem. 1963, 41, 1650; Santos, E.; Rosillo, I.; Del Castillo, B.; Avendãno, C.; J. Chem. Res. 1982, 131.
34. Fujioka, H.; Tari, T.; J. Pharm. Dyn. 1982, 5, 475.
35. Bausch, M. J.; David, B.; Dobrowolski, P.; Prasad, V.; J. Org. Chem. 1990, 55, 5806; Bausch, M. J.; Selmarten, D.; Gostowski, R.; Dobrowolski, P.; J. Phys. Org. Chem. 1991, 4, 67.
36. Kleinpeter, E.; Heyenreich, M.; Kalder, L.; Koch, A.; Henning, D.; Kempter, G.; Benassi, R.; Taddei, F.; J. Mol. Struct. 1997, 403, 111.
37. Cristiani, F.; Devillanova, F. A.; Diaz, A.; Isaia, F.; Verani, G.; Spectrochim. Acta, Part A 1985, 14, 497.
38. Janssen, M. J.; Recl. Trav. Chim. Pays-Bas. 1960, 79, 454.
39. Derkosch, J.; Monatsh. Chem. 1961, 92, 361.
40. Rutkovskii, V.; Ivin, B. A.; Kirillova, V. A.; Smorygo, N. A.; Sochilin, E. G.; Zh. Obshch. Khim. 1970, 40, 1583.
41. Demoen, J.; Bull. Soc. Chim. Belg. 1966, 75, 524.
42. Fayat, C.; Foucaud, D.; Bull. Soc. Chim. Fr. 1971, 3, 987; Bellanato, J.; Avendãno, C.; Ballesteros, P.; Martinez, M.; Spectrochim. Acta, Part A 1979, 35, 807; Nyquist, R. A.; Fiedler, S. L.; Vib. Spectrosc. 1995, 8, 365; Kleinpeter, E.; Klod, S.; Perjéssy, A.; Samaliková, M.; Synderlata, K.; Susteková, Z.; J. Mol. Struct. 2003, 645, 17.
43. Kimura, T.; Nagao, Y.; Bull. Fac. Tech. Hirsaki Univ. 2003, 5, 11.
44. Montaña González, M. T.; Ariza, J. L. G.; Pino, F.; Villanova, R. G.; Talanta 1978, 25, 331.
45. Elmore, D. T.; J. Chem. Soc. 1958, 3459; Jenssen, K. A.; Nielsen, P. A.; Acta Chem. Scand. 1966, 20, 597; Cogrossi, P.; Spectrochim. Acta, Part A 1972, 28, 855.
46. Tan, S. F.; Ang, K. P.; Fong, Y. F.; J. Chem. Soc. Perkin Trans. 2 1986, 1941.
47. Fresneau, P.; Morand, J. M.; Thomasson, F.; Cussac, M.; Spectrochim. Acta, Part A 1999, 55, 2893.
48. Braun, S.; Kalinowski, H. O.; Berger, S.; 100 and More Basic NMR Experiments: A Practical Course, VCH: Weinheim, 1996.
49. Ösz, E.; Szilágyi, L.; Marton, J.; J. Mol. Struct. 1998, 442, 267.
50. Benassi, G.; Bregulla, A.; Friderich, A.; Henning, D.; Heydenreich, M.; Mickler, W.; Kleinpeter E.; Kempter, G.; Schilde, U.; Taddei, F.; J. Mol. Struct. 1998, 441, 47.
51. Santos, M. V. P.; Silva Júnior, M. R.; Oliveira, S. M.; da Silva, J. B. P.; Lima, M. T. C.; Lima, M. C. A.; Galdino, S. L.; Pitta, R.; J. Mol. Struc. Theochem. 2005, 715, 191.
52. Benassi, G.; Bregulla, A.; Henning, D.; Heydenreich, M.; Kempter, G., Kleinpeter E.; Taddei, F.; J. Mol. Struct. 1999, 475, 105.
53. Lempert, K.; Nyitrai, J.; Zaner, K.; Kalman, A.; Argay, Gy.; Duisemberg, A. J. M.; Sohar, P.; Tetrahedron 1973, 29, 3565.
54. Walker, L.A.; Folting, K.;Merrit, L.L.; Acta Crystallogr., Sect. B: Struct. Sci. 1969, 25, 88.
55. Fujiwara, H.; van Der Veen, J. M.; J. Chem. Soc. Perkin Trans. 2 1979, 5, 69.
56. Cassady, R. E.; Hawkinson, S. W.; Acta Crystallogr., Sect. B: Struct. Sci. 1982, 38, 1646.
57. Devillanova, F. A.; Isaía, F.; Verani, G.; Battaglia, L. P.; Corradi, A. B.; J. Chem. Res. 1987, 192.
58. Drew, M. G. B.; Mok, K. F.; Ang, K. P.; Tan, S. F.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 1987, 43, 745.
59. De Simone, C. A.; Zukerman-Schpector, J.; Pereira, M. A.; Luu-Duc, C.; Pitta, I. R.; Galdino, S. L.; Amorim, E. L. C.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 1995, 51, 2620 .
60. De Simone, C. A.; Pereira, M. A.; Luu-Duc, C.; Pitta, I. R.; Galdino, S. L.; Menezes, E. H. C.; Zukerman-Schpector, J.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 1996, 52, 2771.
61. Yu, F. L.; Schwalbe, C. H.; Watkin, D. J.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 2004, 60, 714.
62. Sauli, M.; Ger. Offen. 1972, 2, 923.
63. Marton, J.; Enisz, J.; Hosztafi, S.; Tímàr, T.; J. Agric. Food Chem. 1993, 41, 148.
64. Góes, A. J. S.; Lima, M. C. A.; Galdino, S. L.; Pitta, I. R.; Luu-Duc, C.; J. Pharm. Belg. 1991, 46, 236.
65. Lima, M. C. A.; Costa, L. B.; Góes, A. J. S.; Galdino, S. L.; Pitta, I. R.; Luu-Duc, C.; Pharmazie 1992, 47, 182.
66. Cavalcanti Amorim, E. L.; Ferreira Brandão, S. S.; Morais Cavalcanti, C. O.; Lins Galdino, S.; Rocha Pitta, I.; Luu-Duuc, C.; Ann. Pharm. Fr. 1992, 2, 103.
67. Brandão, S. S. F.; Rocha Filho, J. A.; Chantegrel, J.; Albuquerque, J. F. C.; Ximenes, E. A.; Galdino, S. L.; Pitta, I. R.; Perrissin, M.; Luu-Duuc, C.; Ann. Pharm. Fr. 1997, 55, 206.
68. Albuquerque, J. F. C.; Rocha Filho, J. A.; Brandão, S. S. F.; Lima, M. C. A.; Ximenes, E. A.; Galdino, S. L.; Pitta, I. R.; Chantegrel, J.; Perrissin, M.; Luu-Duuc, C.; Il Fármaco 1999, 54, 77.
69. Henze, H. R.; Smith, P. E.; J. Am. Chem. Soc. 1943, 65, 1090.
70. Singh, S. P.; Auyong, T. K.; Parmar, S. S.; J. Pharm. Sci. 1974, 63, 960.
71. Cortes, S.; Liao, Z. K.; Watson, D.; Kohn, H.; J. Med. Chem. 1985, 28, 601.
72. Danielson, B.; Sundell, L.; Acta. Pharm. Suecia 1965, 2, 137.
73. Ozkirimli, S.; Hamali, O.; Il Farmaco 1995, 50, 65.
74. Karali, N.; Gürsoy, A.; Terzioglu, N.; Özkirimli, S.; Özer, H.; Ekinci, A. C. A.; Arch. Pharm. Med. Chem. 1998, 331, 254.
75. Hardman, J. G.; Limbird, L. E.; Molinoff, P. B.; Ruddon, R. W.; Gilman, A. G.; Goodman & Gilman As Bases Farmacológicas da Terapêutica, 9ª ed., McGraw-Hill: México, 1996.
76. WHO; Model List of Essential Drugs (EDL), 10ª ed., 1997.
77. BRASIL, Portaria nº 1.587/GM do Ministro de Estado da Saúde; Relação Nacional de Medicamentos Essenciais, RENAME, 2002.
78. Scholl, S.; Koch, A.; Henning, D.; Kempter, G.; Kleinpeter, E.; Struct. Chem. 1999, 10, 355.
79. Zha, C.; Brown, G. B.; Brouillete, W. J.; J. Med. Chem. 2004, 47, 6519.
80. Khodorov, B. I.; Biophys. Mol. Biol. 1981, 37, 49.
81. Kanyonyo, M.; Govaerts, S. J.; Hermans, E.; Poupaert, J. H.; Lambert, D. M.; Bioorg. Med. Chem. Lett. 1999, 9, 2233.
82. Muccioli, G. G.; Martin, D.; Scriba, G. K. E.; Poppitz, W.; Cannata, F.; Poupaert, J. H.; Wouters, J.; Lambert, D. M.; J. Med. Chem. 2005, 48, 1748.
83. Muccioli, G. G.; Fazio, N.; Scriba, G. K. E.; Poppitz, W.; Cannata, F.; Poupaert, J. H.; Wouters, J.; Lambert, D. M.; J. Med. Chem. 2006, 49, 417.
84. Luttermoser, G. W.; Bond, H. W.; J. Parasitol. 1954, 40, 33.
85. Benazet, F.; Leroy, J. P.; Bull. Soc. Pat. E.1974, 3, 287; Werbel, L. M.; Elslager, E. F.; J. Med. Chem. 1977, 20, 1569.
86. Oliveira, S. M.; Albuquerque, M. C. P. A.; Pitta, M. G. R.; Malagueño, E.; Santana, J. V.;Lima, M. C. A.; Pitta, I. R.; Galdino, S. L.; Acta Farm. Bonaer. 2004, 23, 343.
87. Albuquerque, M. C. P. A.; Silva, T. G.; Pitta, M. G. R.; Silva, A. C. A.; Silva, P. G.; Malagueño, E.; Santana, J. V.; Wanderley, A. G.; Lima, M. C. A.; Galdino, S. L.; Barbe, J.; Pitta, I. R.; Pharmazie 2005, 60, 13.
88. Know, S. K.; Park, M. S.; J. Med. Chem. 1991, 34, 1845; Santos, L. C.; Mourão, R. H. V.; Uchoa, F. T.; Silva, T. G.; Malta, D. J. N.; Moura, R. O.; Lima, M. C. A.; Galdino, S. L.; Pitta, I. R.; Barbe, J.; J. Heterocyclic Comm. 2005, 11, 433.
89. Elokdah, H.; Sulkowski, T. S.; Abou-Gharbia, M.; Butera, J. A.; Chai, S. Y.; McFarlane, G. R.; McKean, M. L.; Babiak, J. L; Adelman, S. J.; Quinet, E. M.; J. Med. Chem. 2004, 47, 681.
90. Barraclough, P.; Brockwell, M.; Cadwell, A. G.; Demaine, D. A.; Harris, C. J.; King, W. R.; Stepney, R. J.; Wharton, C. J.; Whittle, B. J. R.; Arch. Pharm. - Weinheim. 1994, 327, 307; Barraclough, P.; Bolofo, M. L.; Giles, H.; Gillam, J.; Harris, C. J.; Kelly, M. G.; Leff, P.; McNeill, A.; Robertson, A. D.; Stepney, R. J.; Whittle, B. J. R.; Bioorg. Med. Chem. 1996, 4, 81; Stilz, H. U.; Jablonka, B.; Just, M.; Knolle, J.; Paulus, E. F.; Zoller, G.; J. Med. Chem. 1996, 39, 2118.
91. Dylag, T.; Zygmunt, M.; Maciag, D.; Handzlik, J.; Bednarski, M.; Filipek, B.; Kiec-Kononowicz, K.; Eur. J. Med. Chem. 2004, 39, 1013; Pekala, E.; Stadnika, K.; Broda, A.; Zygmunt, M.; Filipek, B.; Kiec-Kononowicz, K.; Eur. J. Med. Chem. 2005, 40, 259.
92. Kushev, D.; Naydenova, E.; Popova, J.; Maneva, L.; Grancharov, K.; Spassovska, N.; Z. Naturforsch., C: J. Biosci. 2003, 58, 103. Bakalova, A.; Buyukliev, R.; Tcholakova, I.; Momekov, G.; Konstatinov, S.; Karaivanova, M.; Eur. J. Med. Chem. 2005, 38, 627; Opacic, N.; Barbaric, M.; Zorc, B.; Cetina, M.; Nagl, A.; Frkovic, D.; Kralj, M.; Pavelic, M.; Balzarani, J.; Andrei, G.; Snoeck, R.; De Clercq, E.; Raic-Malic, S.; Mintas, M.; J. Med. Chem. 2005, 48, 475.
93. Verlinden, Y.; Cuconati, A.; Wimmer, E.; Rombaut, B.; Antiviral Res. 2000, 48, 61.
94. Lima, J. G.; Galdino, S. L.; Pitta, R.; Catanho, M. T. J.; Perrissin, M.; Luu-Duuc, C.; Ann. Pharm. Fr. 1993, 51, 283.
95. Ooms, F.; Wouters, J.; Oscari, O.; Happaerts, T.; Bouchard, G.; Carrupt, P. A.; Testa, B.; Lambert, D. M.; J. Med. Chem. 2002, 45, 1748; Sutherland, J. J.; Weaver, D. F.; J. Chem. Inf. Comput. Sci. 2003, 43, 1028; Muccioli, G. G.; Wouters, J.; Charlier, C.; Scriba, G. K. E.; Pizza, T.; Di Pace, P.; De Martino, P.; Poppitz, W. F.; Poupaert, J. H.; Lambert, D. M.; J. Med. Chem. 2006, 49, 872.
Recebido em 27/9/06; aceito em 10/8/07; publicado na web em 19/3/08
- 1. Thomas, G.; Química Medicinal Uma Introdução, 1Ş ed., Guanabara Koogan: Rio de Janeiro, 2003.
- 2. Ellestad, G. A.; J. Med. Chem 2006, 49, 6627;
- Barreiro, E. J.; Fraga, C. A. M.; Química Medicinal - As Bases Moleculares da Ação dos Fármacos, 1Ş ed., Artmed: Porto Alegre, 2001.
- 3. Baeyer, A.; Ann. Chem. 1861, 117, 178;
- Finkbeiner, H.; J. Org. Chem. 1965, 30, 3414.
- 4. Klason, P.; Chem. Ztg. 1890, 14, 543.
- 5. Komatsu, S.; Mem. Coll. Sci. Eng. Kyoto Imp. Univ. 1911, 3, 1;
- Wheeler, H. L.; Nicolet, B. H. ; Johnson, T. B.; Am. Chem. J. 1911, 46, 456;
- Johnson, T. B.; Nicolet, B. H.; J. Am. Chem. Soc. 1911, 33, 1975.
- 6. Johnson, T. B.; Chernoff, L. H.; J. Am. Chem. Soc. 1912, 34, 1208.
- 7. Jacobson, R. A.; U.S. Pat. 2 143, 186 1988
- 8. Ware, E.; Chem. Rev. 1950, 46, 403.
- 9. Edward, J. T.; Chem. Org. Sulfur. Comp. Eng. 1966, 2, 287.
- 10. Rogawski, M.; Porter, R. J.; Pharmacol. Rev. 1990, 42, 223.
- 11. Meusel, M.; Gütschow, M.; Org. Prep. Proced. Int. 2004, 36, 391.
- 12. Gaudio, A. C.; Quim. Nova 1995, 19, 278.
- 13. López, C. A.; Trigo, G.; Adv. Heterocyclic Chemistry 1985, 38, 177.
- 14. Edward, J. T.; Liu, J. K.; Can. J. Chem. 1969, 47, 1117.
- 15. Congdon, W. I.; Edward, J. T.; Can. J. Chem 1972, 50, 3767.
- 16. Edward, J. T.; Wong, S.; Can. J. Chem. 1980, 57, 15.
- 17. Nyitrai, J.; Acta Chim. Acad. Sci. Hung. 1967, 51, 95;
- Markovits-Komis, R.; Nyitrai, J.; Lempert, K.; Chem. Ber. 1971, 104, 3080.
- 18. Fetter, J.; Nyitrai, J.; Lempert, K.; Tetrahedron 1971, 27, 5953.
- 19. Orazi, O. O.; Corral, R. A.; Experientia 1965, 21, 508.
- 20. Orazi, O. O.; Corral, R. A.; Schttenberg, H.; J. Chem. Soc. Perkin Trans. 2 1974, 219.
- 21. Trigo, G. G.; Avendaño, C.; Varela, I.; Martinez, M.; An. Quim. 1978, 74, 1090;
- Pedregal, C.; Trigo, G. G.; Espada, M.; Elguero, J.; J. Heterocycl. Chem. 1984, 21, 477.
- 22. Carrington, H. C.; Waring, W. S.; J. Chem. Soc. 1950, 354.
- 23. Edward, J. T.; Liu, J. K.; Can. J. Chem. 1972, 50, 2423.
- 24. Wheeler, L.; Hoffman, C.; Am. Chem. J. 1911, 45, 368.
- 25. Phillips, A. P.; Murphy, J. G.; J. Org. Chem. 1951, 16, 954.
- 26. Baranov, S. N.; Komaritsa, I. D.; Khim. Geterosikl. Soedin.1965, 1, 69.
- 27. Moriya, T.; Hagio, K.; Yoneda, N.; Chem. Pharm. Bull. 1980, 28, 1891.
- 28. Vogels, G. D.; Windt, F. E.; Bassi, W.; Recl. Trav. Chim. Pays. Bas. 1969, 88, 940;
- Ivin, B. A.; Rutkowskii, G. V.; Sochilin, E. G.; Tsereteli, Y. U.; Zhr. Org. Khim. 1972, 8, 840;
- Ivin, B. A.; Rutkowskii, G. V.; Andreev, S. A.; Sochilin, E. G.; Zhr. Org. Khim. 1973, 9, 2194;
- Ivin, B. A.; Rutkowskii, G. V.; Rusavskaya, T. N.; Sochilin, E. G.; Zhr. Org. Khim. 1975, 11, 2188;
- Blagoeva, I. B.; Pojarlieff, I. G.; Dimitrinov, V. S.; J. Chem. Soc. Perkin Trans. 2 1978, 887.
- 29. Edward, J. T.; Nielsen, S.; J. Chem. Soc. 1957, 5084.
- 30. Rakowitz, D.; Maccari, R.; Ottanà, R.; Vigorita, M.G.; Bioorg. Med. Chem 2006, 14, 567.
- 31. Zief, M.; Edsall, J. T.; J. Am. Chem. Soc. 1937, 59, 2245;
- Pickety, L. W.; MClean, M.; J. Am. Chem. Soc. 1939, 61, 423.
- 32. Stuckey, R. E.; J. Chem. Soc. 1947, 331.
- 33. Edward, J. T.; Chin, O. J.; Can. J. Chem. 1963, 41, 1650;
- Santos, E.; Rosillo, I.; Del Castillo, B.; Avendãno, C.; J. Chem. Res. 1982, 131.
- 34. Fujioka, H.; Tari, T.; J. Pharm. Dyn. 1982, 5, 475.
- 35. Bausch, M. J.; David, B.; Dobrowolski, P.; Prasad, V.; J. Org. Chem. 1990, 55, 5806;
- Bausch, M. J.; Selmarten, D.; Gostowski, R.; Dobrowolski, P.; J. Phys. Org. Chem. 1991, 4, 67.
- 36. Kleinpeter, E.; Heyenreich, M.; Kalder, L.; Koch, A.; Henning, D.; Kempter, G.; Benassi, R.; Taddei, F.; J. Mol. Struct. 1997, 403, 111.
- 37. Cristiani, F.; Devillanova, F. A.; Diaz, A.; Isaia, F.; Verani, G.; Spectrochim. Acta, Part A 1985, 14, 497.
- 38. Janssen, M. J.; Recl. Trav. Chim. Pays-Bas. 1960, 79, 454.
- 39. Derkosch, J.; Monatsh. Chem. 1961, 92, 361.
- 40. Rutkovskii, V.; Ivin, B. A.; Kirillova, V. A.; Smorygo, N. A.; Sochilin, E. G.; Zh. Obshch. Khim. 1970, 40, 1583.
- 41. Demoen, J.; Bull. Soc. Chim. Belg. 1966, 75, 524.
- 42. Fayat, C.; Foucaud, D.; Bull. Soc. Chim. Fr. 1971, 3, 987;
- Bellanato, J.; Avendãno, C.; Ballesteros, P.; Martinez, M.; Spectrochim. Acta, Part A 1979, 35, 807;
- Nyquist, R. A.; Fiedler, S. L.; Vib. Spectrosc. 1995, 8, 365;
- Kleinpeter, E.; Klod, S.; Perjéssy, A.; Samaliková, M.; Synderlata, K.; Susteková, Z.; J. Mol. Struct 2003, 645, 17.
- 43. Kimura, T.; Nagao, Y.; Bull. Fac. Tech. Hirsaki Univ. 2003, 5, 11.
- 44. Montaña González, M. T.; Ariza, J. L. G.; Pino, F.; Villanova, R. G.; Talanta 1978, 25, 331.
- 45. Elmore, D. T.; J. Chem. Soc. 1958, 3459;
- Jenssen, K. A.; Nielsen, P. A.; Acta Chem. Scand. 1966, 20, 597;
- Cogrossi, P.; Spectrochim. Acta, Part A 1972, 28, 855.
- 46. Tan, S. F.; Ang, K. P.; Fong, Y. F.; J. Chem. Soc. Perkin Trans. 2 1986, 1941.
- 47. Fresneau, P.; Morand, J. M.; Thomasson, F.; Cussac, M.; Spectrochim. Acta, Part A 1999, 55, 2893.
- 48. Braun, S.; Kalinowski, H. O.; Berger, S.; 100 and More Basic NMR Experiments: A Practical Course, VCH: Weinheim, 1996.
- 49. Ösz, E.; Szilágyi, L.; Marton, J.; J. Mol. Struct. 1998, 442, 267.
- 50. Benassi, G.; Bregulla, A.; Friderich, A.; Henning, D.; Heydenreich, M.; Mickler, W.; Kleinpeter E.; Kempter, G.; Schilde, U.; Taddei, F.; J. Mol. Struct 1998, 441, 47.
- 51. Santos, M. V. P.; Silva Júnior, M. R.; Oliveira, S. M.; da Silva, J. B. P.; Lima, M. T. C.; Lima, M. C. A.; Galdino, S. L.; Pitta, R.; J. Mol. Struc. Theochem. 2005, 715, 191.
- 52. Benassi, G.; Bregulla, A.; Henning, D.; Heydenreich, M.; Kempter, G., Kleinpeter E.; Taddei, F.; J. Mol. Struct 1999, 475, 105.
- 53. Lempert, K.; Nyitrai, J.; Zaner, K.; Kalman, A.; Argay, Gy.; Duisemberg, A. J. M.; Sohar, P.; Tetrahedron 1973, 29, 3565.
- 54. Walker, L.A.; Folting, K.;Merrit, L.L.; Acta Crystallogr., Sect. B: Struct. Sci. 1969, 25, 88.
- 55. Fujiwara, H.; van Der Veen, J. M.; J. Chem. Soc. Perkin Trans. 2 1979, 5, 69.
- 56. Cassady, R. E.; Hawkinson, S. W.; Acta Crystallogr., Sect. B: Struct. Sci. 1982, 38, 1646.
- 57. Devillanova, F. A.; Isaía, F.; Verani, G.; Battaglia, L. P.; Corradi, A. B.; J. Chem. Res. 1987, 192.
- 58. Drew, M. G. B.; Mok, K. F.; Ang, K. P.; Tan, S. F.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 1987, 43, 745.
- 59. De Simone, C. A.; Zukerman-Schpector, J.; Pereira, M. A.; Luu-Duc, C.; Pitta, I. R.; Galdino, S. L.; Amorim, E. L. C.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem 1995, 51, 2620 .
- 60. De Simone, C. A.; Pereira, M. A.; Luu-Duc, C.; Pitta, I. R.; Galdino, S. L.; Menezes, E. H. C.; Zukerman-Schpector, J.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 1996, 52, 2771.
- 61. Yu, F. L.; Schwalbe, C. H.; Watkin, D. J.; Acta Crystallogr., Sect. C: Cryst. Struct. Chem. 2004, 60, 714.
- 62. Sauli, M.; Ger. Offen. 1972, 2, 923.
- 63. Marton, J.; Enisz, J.; Hosztafi, S.; Tímàr, T.; J. Agric. Food Chem. 1993, 41, 148.
- 64. Góes, A. J. S.; Lima, M. C. A.; Galdino, S. L.; Pitta, I. R.; Luu-Duc, C.; J. Pharm. Belg. 1991, 46, 236.
- 65. Lima, M. C. A.; Costa, L. B.; Góes, A. J. S.; Galdino, S. L.; Pitta, I. R.; Luu-Duc, C.; Pharmazie 1992, 47, 182.
- 66. Cavalcanti Amorim, E. L.; Ferreira Brandão, S. S.; Morais Cavalcanti, C. O.; Lins Galdino, S.; Rocha Pitta, I.; Luu-Duuc, C.; Ann. Pharm. Fr. 1992, 2, 103.
- 67. Brandão, S. S. F.; Rocha Filho, J. A.; Chantegrel, J.; Albuquerque, J. F. C.; Ximenes, E. A.; Galdino, S. L.; Pitta, I. R.; Perrissin, M.; Luu-Duuc, C.; Ann. Pharm. Fr. 1997, 55, 206.
- 68. Albuquerque, J. F. C.; Rocha Filho, J. A.; Brandão, S. S. F.; Lima, M. C. A.; Ximenes, E. A.; Galdino, S. L.; Pitta, I. R.; Chantegrel, J.; Perrissin, M.; Luu-Duuc, C.; Il Fármaco 1999, 54, 77.
- 69. Henze, H. R.; Smith, P. E.; J. Am. Chem. Soc. 1943, 65, 1090.
- 70. Singh, S. P.; Auyong, T. K.; Parmar, S. S.; J. Pharm. Sci. 1974, 63, 960.
- 71. Cortes, S.; Liao, Z. K.; Watson, D.; Kohn, H.; J. Med. Chem. 1985, 28, 601.
- 72. Danielson, B.; Sundell, L.; Acta. Pharm. Suecia 1965, 2, 137.
- 73. Ozkirimli, S.; Hamali, O.; Il Farmaco 1995, 50, 65.
- 74. Karali, N.; Gürsoy, A.; Terzioglu, N.; Özkirimli, S.; Özer, H.; Ekinci, A. C. A.; Arch. Pharm. Med. Chem. 1998, 331, 254.
- 75. Hardman, J. G.; Limbird, L. E.; Molinoff, P. B.; Ruddon, R. W.; Gilman, A. G.; Goodman & Gilman As Bases Farmacológicas da Terapêutica, 9Ş ed., McGraw-Hill: México, 1996.
- 76. WHO; Model List of Essential Drugs (EDL), 10Ş ed., 1997.
- 77. BRASIL, Portaria nº 1.587/GM do Ministro de Estado da Saúde; Relação Nacional de Medicamentos Essenciais, RENAME, 2002.
- 78. Scholl, S.; Koch, A.; Henning, D.; Kempter, G.; Kleinpeter, E.; Struct. Chem. 1999, 10, 355.
- 79. Zha, C.; Brown, G. B.; Brouillete, W. J.; J. Med. Chem. 2004, 47, 6519.
- 80. Khodorov, B. I.; Biophys. Mol. Biol. 1981, 37, 49.
- 81. Kanyonyo, M.; Govaerts, S. J.; Hermans, E.; Poupaert, J. H.; Lambert, D. M.; Bioorg. Med. Chem. Lett. 1999, 9, 2233.
- 82. Muccioli, G. G.; Martin, D.; Scriba, G. K. E.; Poppitz, W.; Cannata, F.; Poupaert, J. H.; Wouters, J.; Lambert, D. M.; J. Med. Chem. 2005, 48, 1748.
- 83. Muccioli, G. G.; Fazio, N.; Scriba, G. K. E.; Poppitz, W.; Cannata, F.; Poupaert, J. H.; Wouters, J.; Lambert, D. M.; J. Med. Chem. 2006, 49, 417.
- 84. Luttermoser, G. W.; Bond, H. W.; J. Parasitol. 1954, 40, 33.
- 85. Benazet, F.; Leroy, J. P.; Bull. Soc. Pat. E.1974, 3, 287;
- Werbel, L. M.; Elslager, E. F.; J. Med. Chem 1977, 20, 1569.
- 86. Oliveira, S. M.; Albuquerque, M. C. P. A.; Pitta, M. G. R.; Malagueño, E.; Santana, J. V.;Lima, M. C. A.; Pitta, I. R.; Galdino, S. L.; Acta Farm. Bonaer. 2004, 23, 343.
- 87. Albuquerque, M. C. P. A.; Silva, T. G.; Pitta, M. G. R.; Silva, A. C. A.; Silva, P. G.; Malagueño, E.; Santana, J. V.; Wanderley, A. G.; Lima, M. C. A.; Galdino, S. L.; Barbe, J.; Pitta, I. R.; Pharmazie 2005, 60, 13.
- 88. Know, S. K.; Park, M. S.; J. Med. Chem. 1991, 34, 1845;
- Santos, L. C.; Mourão, R. H. V.; Uchoa, F. T.; Silva, T. G.; Malta, D. J. N.; Moura, R. O.; Lima, M. C. A.; Galdino, S. L.; Pitta, I. R.; Barbe, J.; J. Heterocyclic Comm. 2005, 11, 433.
- 89. Elokdah, H.; Sulkowski, T. S.; Abou-Gharbia, M.; Butera, J. A.; Chai, S. Y.; McFarlane, G. R.; McKean, M. L.; Babiak, J. L; Adelman, S. J.; Quinet, E. M.; J. Med. Chem. 2004, 47, 681.
- 90. Barraclough, P.; Brockwell, M.; Cadwell, A. G.; Demaine, D. A.; Harris, C. J.; King, W. R.; Stepney, R. J.; Wharton, C. J.; Whittle, B. J. R.; Arch. Pharm. - Weinheim. 1994, 327, 307;
- Barraclough, P.; Bolofo, M. L.; Giles, H.; Gillam, J.; Harris, C. J.; Kelly, M. G.; Leff, P.; McNeill, A.; Robertson, A. D.; Stepney, R. J.; Whittle, B. J. R.; Bioorg. Med. Chem. 1996, 4, 81;
- Stilz, H. U.; Jablonka, B.; Just, M.; Knolle, J.; Paulus, E. F.; Zoller, G.; J. Med. Chem. 1996, 39, 2118.
- 91. Dylag, T.; Zygmunt, M.; Maciag, D.; Handzlik, J.; Bednarski, M.; Filipek, B.; Kiec-Kononowicz, K.; Eur. J. Med. Chem. 2004, 39, 1013;
- Pekala, E.; Stadnika, K.; Broda, A.; Zygmunt, M.; Filipek, B.; Kiec-Kononowicz, K.; Eur. J. Med. Chem. 2005, 40, 259.
- 92. Kushev, D.; Naydenova, E.; Popova, J.; Maneva, L.; Grancharov, K.; Spassovska, N.; Z. Naturforsch., C: J. Biosci. 2003, 58, 103.
- Bakalova, A.; Buyukliev, R.; Tcholakova, I.; Momekov, G.; Konstatinov, S.; Karaivanova, M.; Eur. J. Med. Chem. 2005, 38, 627;
- Opacic, N.; Barbaric, M.; Zorc, B.; Cetina, M.; Nagl, A.; Frkovic, D.; Kralj, M.; Pavelic, M.; Balzarani, J.; Andrei, G.; Snoeck, R.; De Clercq, E.; Raic-Malic, S.; Mintas, M.; J. Med. Chem. 2005, 48, 475.
- 93. Verlinden, Y.; Cuconati, A.; Wimmer, E.; Rombaut, B.; Antiviral Res. 2000, 48, 61.
- 94. Lima, J. G.; Galdino, S. L.; Pitta, R.; Catanho, M. T. J.; Perrissin, M.; Luu-Duuc, C.; Ann. Pharm. Fr. 1993, 51, 283.
- 95. Ooms, F.; Wouters, J.; Oscari, O.; Happaerts, T.; Bouchard, G.; Carrupt, P. A.; Testa, B.; Lambert, D. M.; J. Med. Chem. 2002, 45, 1748;
- Sutherland, J. J.; Weaver, D. F.; J. Chem. Inf. Comput. Sci. 2003, 43, 1028;
- Muccioli, G. G.; Wouters, J.; Charlier, C.; Scriba, G. K. E.; Pizza, T.; Di Pace, P.; De Martino, P.; Poppitz, W. F.; Poupaert, J. H.; Lambert, D. M.; J. Med. Chem. 2006, 49, 872.
Datas de Publicação
-
Publicação nesta coleção
16 Jun 2008 -
Data do Fascículo
2008
Histórico
-
Aceito
10 Ago 2007 -
Recebido
27 Set 2006