Resumo
Chromatography combined with several different detection systems is one of the more used and better performing analytical tools. Chromatography with tandem mass spectrometric detection gives highly selective and sensitive analyses and permits obtaining structural information about the analites and about their molar masses. Due to these characteristics, this analytical technique is very efficient when used to detect substances at trace levels in complex matrices. In this paper we review instrumental and technical aspects of chromatography-tandem mass spectrometry and the state of the art of the technique as it is applied to analysis of toxic substances in food.
tandem mass spectrometry; chromatography; food
tandem mass spectrometry; chromatography; food
REVISÃO
O estado da arte da cromatografia associada à espectrometria de massas acoplada à espectrometria de massas na análise de compostos tóxicos em alimentos
The state of the art of chromatography associated with the tandem mass spectrometry for toxic compound analyses in food
Mariza C. Chiaradia* * e-mail: chiaradia@iqm.unicamp.br ; Carol H. Collins; Isabel C. S. F. Jardim
Departamento de Química Analítica, Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas SP, Brasil
ABSTRACT
Chromatography combined with several different detection systems is one of the more used and better performing analytical tools. Chromatography with tandem mass spectrometric detection gives highly selective and sensitive analyses and permits obtaining structural information about the analites and about their molar masses. Due to these characteristics, this analytical technique is very efficient when used to detect substances at trace levels in complex matrices. In this paper we review instrumental and technical aspects of chromatography-tandem mass spectrometry and the state of the art of the technique as it is applied to analysis of toxic substances in food.
Keywords: tandem mass spectrometry; chromatography; food.
INTRODUÇÃO
A cromatografia pode ser combinada a diferentes sistemas de detecção, tratando-se de uma das técnicas analíticas mais utilizadas e de melhor desempenho. O acoplamento de um cromatógrafo com o espectrômetro de massas combina as vantagens da cromatografia (alta seletividade e eficiência de separação) com as vantagens da espectrometria de massas (obtenção de informação estrutural, massa molar e aumento adicional da seletividade)1. Para que o acoplamento seja possível, idealmente, é necessário que as características de cada instrumento não sejam afetadas pela sua conexão, assim como não devem ocorrer modificações químicas não controladas do analito e perda de amostra durante a sua passagem do cromatógrafo para o espectrômetro de massas2.
As técnicas cromatográficas mais comumente acopladas à espectrometria de massas ("mass spectrometry") - EM são a cromatografia gasosa ("gas chromatography") - CG e a cromatografia líquida de alta eficiência ("high performance liquid chromatography") - CLAE. A combinação com outras técnicas de separação, como a eletroforese capilar, a cromatografia em camada delgada e a cromatografia de permeação em gel é possível, mas usada com menor freqüência.
CROMATOGRAFIA GASOSA ACOPLADA À ESPECTROMETRIA DE MASSAS (CG-EM)
A combinação da cromatografia gasosa com a espectrometria de massas é relativamente simples, uma vez que as características de funcionamento do cromatógrafo a gás são suficientemente compatíveis com a necessidade de alto vácuo do espectrômetro de massas2. Quando são utilizadas colunas capilares em CG é possível conectar a saída da coluna diretamente à fonte do espectrômetro, uma vez que, em condições normais de operação, o sistema de bombeamento do espectrômetro de massas é capaz de captar todo o eluente da coluna3. Quando são utilizadas colunas recheadas, a vazão do eluente deve ser reduzida antes da sua entrada na fonte de ionização do espectrômetro. Para isto, podem ser utilizados divisores de fluxo, mas seu desempenho não é tão satisfatório, uma vez que podem gerar perdas na detectabilidade3. O interfaceamento, neste caso, pode ser feito por meio de um separador de jato, que é constituído por dois capilares alinhados e separados por uma distância de cerca de 1 mm, tendo vácuo estabelecido entre eles por meio de uma bomba. Desta maneira, quando o eluente da coluna atravessa o primeiro capilar, grande parte do gás carregador (geralmente He), que possui baixa massa atômica, é bombeado para fora dos capilares, enquanto as moléculas de analito (com maior massa) mantêm-se em fluxo linear e penetram no segundo capilar que as direciona para a fonte de íons do espectrômetro de massas3.
Os métodos de ionização mais empregados em CG-EM são ionização por impacto de elétrons ("electron ionization") - IE e a ionização química ("chemical ionization") - IQ1,3. Na IE o analito de interesse, em fase gasosa, é bombardeado com elétrons de alta energia (geralmente 70 eV). As moléculas do analito absorvem esta energia desencadeando vários processos, dentre os quais o mais simples é aquele em que o analito é ionizado pela remoção de um único elétron (M+). Este processo requer tipicamente 10 eV e o restante da energia gera fragmentação dos analitos2,3. Isto consiste em um dos maiores problemas encontrados na aplicação de IE, pois a fragmentação rápida pode conduzir a não observação do íon molecular no espectro, perdendo-se, portanto, uma das mais importantes informações analíticas oferecidas pela EM2.
A IQ é a técnica que foi desenvolvida especialmente para aumentar a produção do íon molecular e reduzir as fragmentações associadas à ionização por elétrons. Nesta técnica, as moléculas do analito, em fase gasosa, são introduzidas na câmara de ionização do espectrômetro de massas, que contém um gás reagente. Esta mistura (moléculas do analito + gás reagente) é bombardeada com elétrons, assim como na IE. Mas, como o gás reagente está em excesso em relação ao analito (geralmente em proporção maior que 1000:1), ele é ionizado quase que exclusivamente e passam a ocorrer reações entre os íons em fase gasosa do gás reagente e as moléculas neutras do analito, dando origem aos íons pseudo-moleculares do analito [M+H]+. Por este processo ser relativamente de baixa energia, quase não é observada fragmentação1,2.
A CG-EM é aplicável a compostos voláteis e termicamente estáveis nas temperaturas relativamente elevadas empregadas durante o processo de separação cromatográfica. Estes requisitos são semelhantes àqueles necessários para que compostos sejam ionizados por meio de IE e IQ2.
CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA ACOPLADA A ESPECTROMETRIA DER MASSAS (CL-EM)
Quando se utiliza a CL-EM, são encontradas incompatibilidades relacionadas à vazão do eluente do sistema cromatográfico com relação à velocidade de bombeamento do sistema de vácuo e o projeto da fonte de íons do espectrômetro de massas1. As vazões utilizadas em CLAE são relativamente grandes (da ordem de 1,0 mL min-1), de maneira que não é possível bombear o eluente de um cromatógrafo a líquido diretamente para o interior da fonte do espectrômetro, que opera a pressões de cerca de 1,3 x 10-4 Pa2. Assim, uma das mais importantes funções de uma interface empregada em CL-EM é remover toda ou, pelo menos, uma parte significativa da fase móvel (FM)2.
Além do problema existente com relação à vazão, os compostos que são separados por CLAE são relativamente pouco voláteis e/ou sensíveis à temperatura, de maneira que não é possível ionizá-los utilizando as técnicas de ionização mais comumente aplicadas na EM2. Desta forma, para o acoplamento da CLAE à EM foi necessário o desenvolvimento de interfaces e de formas de ionização alternativas.
Na tentativa de minimizar os problemas encontrados no interfaceamento do sistema CLAE com EM foram desenvolvidas várias interfaces, nas quais, muitas vezes, também é realizada a ionização do analito por métodos que permitem a obtenção de íons a partir de moléculas sensíveis à temperatura e/ou pouco voláteis2. Por esse motivo muitos autores referem-se a algumas dessas interfaces simplesmente como fontes de ionização. Dentre as fontes de ionização desenvolvidas, as mais empregadas são ionização por eletronebulização ("electrospray ionization") - IEN, ionização química à pressão atmosférica ("atmospheric pressure chemical ionization") - IQPA e, mais recentemente, a fotoionização à pressão atmosférica ("atmospheric pressure photoionization") - FIPA1,4-7.
Em IEN, o líquido no qual o analito de interesse se encontra dissolvido (na FM, no caso do eluente da CLAE) passa através de um capilar, à pressão atmosférica, mantido sob alta voltagem. Na saída do capilar são formadas pequenas gotas altamente carregadas ("spray") que são dessolvatadas ao se deslocarem em sentido contrário ao posicionamento de um eletrodo em uma região de pressão atmosférica. A dessolvatação é assistida por um fluxo contínuo de gás seco (geralmente N2) na região do "spray". À medida que ocorre a dessolvatação, o tamanho das gotas é reduzido até o ponto em que a força de repulsão entre as cargas similares fica maior que as forças de coesão da fase líquida (tensão superficial). Neste momento ocorre a chamada "explosão coulômbica", que gera gotas com tamanhos equivalentes a 10% do tamanho das gotas a partir das quais se originaram. Uma série de explosões passa então a ocorrer até que são produzidos íons do analito a partir destas gotas, os quais são transferidos para o interior do espectrômetro de massas por uma série de dispositivos de focalização1,2,4.
Como em IEN a ionização ocorre diretamente na solução, compostos sensíveis à temperatura podem ser ionizados sem sofrer degradação1,2. Uma vez que em IEN são gerados íons com múltiplas cargas, esta técnica pode ser aplicada a compostos com massas molares relativamente grandes pois, como o espectrômetro de massas mede a razão massa/carga (m/z) dos íons, o intervalo de "massa" de aplicabilidade do instrumento pode ser expandido por um fator equivalente ao número de cargas do íon, isto é, um íon com m/z 1000 e com 20 cargas representa um composto com uma massa molar de 20 000 Da2. Devido ao modo de obtenção dos íons por esta fonte de ionização, a sua aplicação a compostos ionizáveis em solução e compostos altamente polares que podem ser facilmente ionizados é favorecida1.
Uma vez que a IEN é dependente da concentração do analito no eluente da coluna cromatográfica, o uso de divisores de fluxo, para diminuir a vazão do eluente para o interior da interface, não afeta, de forma notável, sua detectabilidade6. Há a necessidade de uso desses divisores de fluxo somente quando a vazão utilizada no cromatógrafo ultrapassar 1 ou 0,5 mL min-1 no caso de FM com elevada porcentagem de água em sua composição1.
Na IQPA, o eluente da coluna cromatográfica passa através de um nebulizador pneumático no qual gotas são geradas e dessolvatadas. O "spray" formado passa por uma região aquecida na qual o vapor é seco, formando espécies neutras que passam através de uma corona de descarga. Um campo suficiente para gerar ionização é aplicado na corona. Como o solvente (proveniente da FM) encontra-se em maior concentração no "spray" que o analito, este é ionizado preferencialmente e passam a ocorrer reações entre estes íons em fase gasosa e as moléculas neutras do analito, o que dá origem aos íons do analito (IQ)1,2,4.
A IQPA pode ser considerada uma fonte de ionização complementar à IEN, pois é aplicável a compostos apolares ou de média polaridade, voláteis e termicamente estáveis, uma vez que a ionização ocorre em fase gasosa2,6 e também porque IQPA é aplicável a vazões de eluente cromatográfico maiores que as suportadas por IEN (0,5 a 2,0 mL min-1)2.
Tanto na IEN quanto na IQPA ocorre a denominada ionização suave, isto é, são formados íons pseudo-moleculares intactos, [M+H]+ no modo positivo ou [M-H]- no modo negativo, porque a energia empregada nestas fontes de ionização não é suficiente para gerar uma fragmentação significativa das moléculas do analito1,2,6. Nestas fontes de ionização, mais comumente na IQPA, também pode ocorrer a formação de adutos entre os íons moleculares e Na+, K+, NH4+, HCOO- e CH3COO-, que são espécies iônicas presentes nas FM provenientes dos modificadores adicionados a elas2,6.
A FIPA é uma técnica de ionização relativamente nova, introduzida por Bruins e colaboradores8 em 2000, que pode ser considerada complementar às outras técnicas de ionização à pressão atmosférica (IEN e IQPA), uma vez que certos grupos de compostos apolares e de baixa polaridade, como hidrocarbonetos policíclicos aromáticos, só podem ser analisados por CL-EM quando se aplica a fotoionização7. A fonte de FIPA é muito semelhante à de IQPA, sendo constituída por um nebulizador aquecido que transforma o eluente da coluna cromatográfica em um "spray" e gera sua dessolvatação parcial. Entretanto, no lugar da corona de descarga da IQPA a FIPA possui uma lâmpada UV com potência de 10,2 eV, cuja função é ocasionar a ionização das moléculas do analito presentes nas gotículas do "spray"7. A detectabilidade em FIPA é comparável à em IQPA, quando são empregadas altas vazões do eluente e é superior, quando utilizadas baixas vazões. Esta fonte de ionização é também menos suscetível à supressão iônica induzida pela matriz e interferências químicas ocasionadas pela presença de tampões no eluente que a IEN e a IQPA7.
A FIPA baseia-se na formação de um íon molecular radical a partir da absorção de um fóton por uma molécula, seguida da ejeção de um elétron, como pode ser observado na Equação 1. Isto só é possível quando a energia do fóton irradiador (hn) é maior que o potencial de ionização (PI) da molécula. Este processo é denominado fotoionização direta7.

A probabilidade de ocorrer a fotoionização direta é baixa, uma vez que os fótons de UV têm uma baixa penetração numa mistura densa de gases e vapores à pressão atmosférica. Entretanto, se for empregada uma alta concentração de uma substância que é facilmente ionizada (um dopante) é possível aumentar significativamente o número de íons gerados pela fotoionização inicial do dopante e subseqüente troca de carga com o analito (Equações 2 e 3, respectivamente), desde que a afinidade eletrônica (AE) do analito seja maior que a do dopante. Mas, se a afinidade protônica (AP) do analito é maior que a AP do íon dopante desprotonado, moléculas do solvente ou clusters podem agir como intermediários entre o íon dopante e o analito pela abstração de um próton do íon dopante (Equação 4), passando este para o analito (Equação 5)7.

Mas a FIPA não é limitada ao modo de ionização positivo. A formação de íons negativos na FIPA é iniciada pela formação de elétrons térmicos durante a fotoionização do solvente (Equação 6) e do gás oxigênio (Equação 7) por meio da transferência de próton (Equações 8 e 9 a,b) ou troca de carga (10), ou ainda por captura de elétons (11) ou reações de substituição (12)7.

onde: X = H, halogênios, NO2.
Portanto, tanto na formação de íons positivos quanto negativos por FIPA, o analito é analisado de forma indireta, por meio de reações químicas foto-iniciadas7.
ESPECTROMETRIA DE MASSAS ACOPLADA À ESPECTROMETRIA DE MASSAS (EM-EM)
A EM-EM é a técnica espectrométrica que, ao invés de utilizar apenas um analisador de massas para separar os íons de mesma razão m/z gerados na fonte de ionização, utiliza dois estágios de espectrometria de massas (MS1 e MS2), em que um deles é usado para isolar o íon de interesse e o outro é usado para estabelecer uma relação entre este íon de interesse isolado e outros íons que foram gerados a partir da sua decomposição induzida2. Esta técnica é amplamente empregada na detecção de compostos presentes em baixas concentrações em matrizes complexas, acoplada à cromatografia, uma vez que possibilita um aumento na detectabilidade e reduz a interferência espectral de compostos presentes na matriz, além de aumentar a quantidade de informação estrutural que se pode obter1.
ANALISADORES DE MASSAS
Quadrupolo
O quadrupolo é constituído de 4 hastes. Os pares opostos das hastes estão conectados eletricamente e uma voltagem com radiofreqüência de 180º fora de fase é aplicada entre eles. Em um valor específico de voltagem, íons de uma determinada razão m/z atravessam o quadrupolo descrevendo uma trajetória estável2. O esquema de um analisador quadrupolar encontra-se na Figura 1.
Analisador de tempo de vôo ("Time-of-Flight Analiser") - ATV
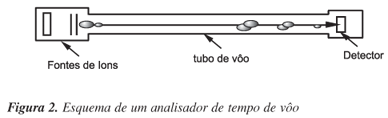
Os ATV baseiam-se no princípio de que, como os íons são gerados na mesma fonte de ionização do espectrômetro de massas, eles possuem a mesma energia cinética, de maneira que as suas velocidades serão apenas diferenciadas pelas suas massas (velocidade é inversamente proporcional à raiz quadrada da massa do íon). Por isto, neste analisador de massas, os íons produzidos na fonte de ionização do espectrômetro são acelerados através de um "tubo de vôo" para serem identificados, uma vez que o tempo que levam para atravessá-lo está relacionado com a razão m/z de cada íon2. Na Figura 2 pode ser visualizado o ATV.
Ion-trap
O ion-trap é um quadrupolo tridimensional que "captura" todos os íons que são introduzidos em seu interior e os mantêm "aprisionados" até que uma determinada radiofreqüência seja aplicada e torna os íons de certa razão m/z instáveis, de forma que são libertados do trap2,9. O analisador de massas ion-trap está mostrado na Figura 3.
ANALISADORES DE MASSAS EM EM-EM
Triplo quadrupolo
Este instrumento é constituído por três quadrupolos em série, sendo que o segundo quadrupolo não é utilizado para separar íons de mesma razão m/z, mas sim como cela de colisão, na qual ocorre a fragmentação dos íons selecionados no primeiro quadrupolo geralmente por dissociação induzida por colisão com um gás inerte ("collision-induced dissociation") - DIC, e também é empregado como direcionador dos íons produzidos ao terceiro quadrupolo.
Na DIC, o íon precursor proveniente do primeiro quadrupolo é acelerado por um potencial elétrico para uma região de alto vácuo no interior do segundo quadrupolo, onde sofre repetidas colisões com um gás inerte de elevada energia (geralmente Ar, He ou N2), o que leva a um aumento na energia potencial deste íon até ocasionar sua fragmentação, conduzindo à formação dos íons produto. Quando DIC é realizada em baixa energia, as reações de fragmentação levam geralmente à perda de fragmentos neutros (H2O, MeOH, CO, CO2 etc.), dependendo da natureza do íon precursor. Esta perda de fragmentos neutros é muito importante na determinação estrutural da molécula do analito, uma vez que fornece informações acerca de grupos funcionais presentes na molécula. Quando DIC é realizada sob elevada energia, as reações de fragmentação geram informações estruturais mais significativas, uma vez que pode levar à quebra das moléculas em posições características. Porém, quando a energia é muito elevada pode levar a uma fragmentação descontrolada10.
Além de informações estruturais, DIC pode melhorar a detectabilidade do método quando usada para gerar um íon característico de uma molécula e assim realizar sua detecção a partir deste íon fragmento quando a molécula do analito de interesse se encontra em presença de outras moléculas de mesma massa molar nominal, uma vez que reduz o ruído e aumenta a detectabilidade.
Todos os quadrupolos são controlados para transmitir íons de uma única razão m/z ou de um intervalo de razões m/z para gerar informação analítica mais exata2,9.
Na Figura 4 encontra-se esquematizado um triplo quadrupolo.
Quadrupolo-ATV (Q-ATV)
É um instrumento em que o último estágio do triplo quadrupolo é substituído por um ATV disposto ortogonalmente ao segundo quadrupolo. Este instrumento, ao invés de varrer seqüencialmente um intervalo de razões m/z selecionadas e gerar o espectro EM-EM das massas selecionadas, detecta todos os íons que entram no tubo de vôo em um tempo específico, o que só é possível devido à sua alta velocidade de varredura2,11. Na Figura 5 pode ser visualizado o analisador Q-ATV.
"Ion-trap"
Neste instrumento a seleção, a decomposição e a subseqüente análise dos íons é realizada na mesma parte do instrumento, de maneira que estes processos ocorrem separados apenas pelo tempo. Para isso, o "trap" é ajustado para capturar todos os íons que entram no espectrômetro de massas e os íons de m/z que não são de interesse são levados à instabilidade para ocasionar sua ejeção do "trap". O(s) íon(s) remanescente(s) no "trap" é(são) dissociado(s). Os íons produzidos na dissociação do íon de m/z selecionado tornam-se seqüencialmente instáveis e são liberados do trap para gerar o espectro de massas2.
TÉCNICAS DE VARREDURA PARA OBTENÇÃO DO ESPECTRO DE MASSAS EM EM-EM2
Varredura dos íons produzidos ("product-ion scan")
Nesta varredura, considerando-se um espectrômetro de massas do tipo triplo quadrupolo, no primeiro estágio (EM1) é isolado o íon de interesse que, em seguida, é fragmentado na cela de colisão. No segundo estágio do espectrômetro de massas (EM2) é feita a varredura dos íons produzidos a partir da fragmentação do íon de interesse isolado em EM1 para obtenção do espectro de massas. Esta técnica também pode ser realizada no "ion-trap".
Varredura do íon precursor ("precursor-ion scan")
No triplo quadrupolo, este tipo de varredura é realizado quando o EM1 é ajustado para transmitir íons dentro de um intervalo de m/z de interesse, os quais são fragmentados na cela de colisão, para que em EM2 sejam transmitidos íons de uma única razão m/z (íon produto de fragmentação). O sinal só é gerado no detector quando EM1 e EM2 estão transmitindo, isto é, quando um íon transmitido por EM1 se fragmenta na cela de colisão gerando o íon produto selecionado que atravessa EM2.
Varredura da constante perda de fragmentos neutros ("constant-neutral-loss scan")
Esta varredura permite observar íons que se fragmentam perdendo uma massa de estrutura química específica e neutra. É realizada no triplo quadrupolo varrendo-se todos os estágios da EM-EM simultaneamente para comparar as diferenças entre as massas varridas por eles, isto é, se em EM1 está sendo transmitido um íon de m/z 100 ao mesmo tempo em que EM2 transmite um íon de m/z 58 e, em seguida, EM1 muda para m/z 101 e EM2 transmite m/z 59, e assim por diante, é porque está havendo uma perda constante de massa neutra de 42 Da.
Monitoramento seletivo de reações ("selected-reaction monitoring") - MSR
Neste tipo de varredura é monitorada a fragmentação de um íon precursor selecionado no triplo quadrupolo por EM1 aos seus correspondentes íons produtos que atravessam EM2. No "ion-trap" o íon precursor é selecionado da mesma forma que na varredura dos íons produzidos mas, neste caso, um íon produto de uma única razão m/z de interesse é confinado.
Quando se monitora a fragmentação de vários íons precursores simultaneamente, este modo de varredura é denominado "monitoramento de reações múltiplas" ("multiple-reaction monitoring") - MRM.
CROMATOGRAMAS DE MASSAS
Quando se faz o acoplamento da cromatografia com a EM obtém-se o chamado cromatograma de massas que é assim denominado por se tratar de um cromatograma constituído de todos os íons produzidos pelo espectrômetro de massas ou apenas pelos íons de interesse produzidos por este12.
O cromatograma contendo todos os íons produzidos pelo espectrômetro de massas é denominado "cromatograma de íons totais" ("total ion chromatogram") - CIT12.
O cromatograma constituído apenas pelos íons de interesse pode ser obtido pelo monitoramento dos íons selecionados ("selected ion monitoring") MIS, ajustando-se o detector de massas para que sejam observados apenas os íons de razão m/z de interesse ou selecionando-os a partir de um banco de dados que contenha os espectros de massas completos12.
UTILIZAÇÃO DA CROMATOGRAFIA COM DETECÇÃO POR ESPECTROMETRIA DE MASSAS ACOPLADA À ESPECTROMETRIA DE MASSAS NA DETERMINAÇÃO DE SUBSTÂNCIAS TÓXICAS EM ALIMENTOS
Substâncias tóxicas são aquelas que impactam negativamente o metabolismo normal dos seres vivos, podendo levar ao estabelecimento de anormalidades fisiológicas e/ou anatômicas13.
A presença de substâncias tóxicas em alimentos pode ser ocasionada por diversas fontes de contaminação. Uma das principais fontes de contaminação de alimentos é a utilização de agroquímicos, que são substâncias empregadas para aumentar a qualidade e a quantidade de alimentos necessários ao sustento da população13. Dentre os agroquímicos utilizados estão os agrotóxicos (herbicidas, inseticidas, fungicidas, acaricidas, fumigantes), que são aplicados a frutas e vegetais em vários estágios de cultivo e pós-colheita, para protegê-los do ataque de pestes e mantê-los próprios para o consumo14. Outro tipo de agroquímico empregado, que pode gerar contaminação de alimentos de origem animal (carne, leite e ovos), são os medicamentos veterinários14.
Além da contaminação por agroquímicos os alimentos também podem ser contaminados por toxinas geradas por microorganismos, como bactérias ou fungos (micotoxinas), por compostos tóxicos produzidos durante o preparo dos alimentos (acrilamidas, aminas heterocíclicas), por compostos presentes na composição de embalagens (estirenos, ftalatos), ou ainda por exposição dos alimentos a contaminantes ambientais (hidrocarbonetos polialogenados, hidrocarbonetos policíclicos aromáticos, organometálicos)6,14. Existem poucos trabalhos na literatura relatando estes dois últimos tipos de contaminação15,16.
Para garantir a segurança da população quanto ao consumo de alimentos, agências governamentais no mundo todo realizam um controle da presença dos contaminantes nos alimentos para que não gerem danos à saúde dos consumidores estabelecendo, para tanto, os limites máximos de resíduos ("maximun residue levels") - LMR6,14,17,18. Para garantir que os LMR sejam respeitados, as metodologias analíticas utilizadas para a determinação de substâncias tóxicas em alimentos devem ser capazes de quantificar resíduos dessas substâncias em concentrações muito baixas, assim como identificá-los de maneira inequívoca. Devido à complexidade das matrizes de alimentos (mistura de água, proteínas, lipídios, carboidratos, vitaminas e minerais14), freqüentemente é necessária uma preparação intensiva da amostra, como também o acoplamento de técnicas analíticas para obtenção de maior seletividade e detectabilidade6.
Neste sentido, mostra-se vantajoso o emprego da cromatografia com detecção por espectrometria de massas acoplada a espectrometria de massas, uma vez que por EM-EM é possível obter uma grande quantidade de informação estrutural acerca do analito, o que assegura sua identificação com maior exatidão do que quando ela é feita apenas com base nas características de retenção dos compostos analisados, como ocorre nas outras técnicas de detecção cromatográficas1. Além disso, quando existem compostos que não podem ser totalmente separados pela técnica cromatográfica empregada, usando EM-EM é possível detectá-los individualmente se possuírem diferentes massas molares ou gerarem diferentes espectros de massas1. Graças a essa elevada seletividade, os efeitos da interferência de componentes da matriz sobre o sinal obtido são minimizados, de forma que procedimentos mais simples de preparo das amostras podem ser empregados, eliminando, muitas vezes, a necessidade de realizar várias etapas de limpeza da amostra ("clean-up"). Isto diminui o custo e o tempo necessários para a realização das análises, de forma que possam ser aplicadas como procedimentos de rotina em laboratórios de controle da qualidade de alimentos19,20. Quando se utiliza a EM-EM é possível obter maior detectabilidade e menores limites de detecção e quantificação do que quando se utiliza EM, devido aos modos de varreduras possíveis de serem realizados, o que favorece a sua aplicação à análise de traços20.
APLICAÇÃO DE CG-EM-EM E CL-EM-EM NA DETERMINAÇÃO DE AGROTÓXICOS EM ALIMENTOS
Arrebola et al.21 desenvolveram um método para determinação de resíduos de agrotóxicos de 81 classes diferentes em vegetais frescos com grande conteúdo de água, como pepino, utilizando CG-EM-EM. Os agrotóxicos analisados foram: acefato, acrinatrina, azoxistrobina, betalaxil, bifentrina, bromopropilato, bupirimato, buprofezina, carbofenotiom, cialotrim, ciflutrina, cipermetrina, ciproconazol, clorfenvinfós, cloropirifós, cloropirifós metílico, clortalonil, clozolinato, deltametrina, diazinona, diclorvós, dicofol, difenoconazol, dimetoato, dissulfotom, a-endossulfam, b-endossulfam, endossulfam-sulfato, esfenvalerato, etiofencarbe, etiona, etoprofós, etrinfós, fenamifós, fenarimol, fenitrotiona, fenpropatrina, fentiona, flucitrinato, fludioxinil, forato, formotiom, fosalona, furatiocarbe, heptenofós, hexaconazol, iprodiona, isofenfós, lindano, malationa, metalaxil, metamidofós, mevinfós, miclobutanil, nuarimol, ometoato, oxadixil, parationa metílico, penconazol, pendimetalina, permetrina, pirazofós, piridabem, pirifenox, pirimetanil, pirimicarbe, pirimifós metílico, piriproxifem, procimidona, propargito, propiconazol, propoxur, quinometionato, tebuconazol, tebufenozide, tetraconazol, tetradifona, triadimefom, triadimenol, triflumizol e vinclozolina.
Para a extração dos agrotóxicos das matrizes de interesse foi utilizado apenas diclorometano e para eliminar a água, sulfato de sódio anidro. O extrato obtido foi redissolvido em cicloexano, seguido pela adição de padrão interno (cafeína) e injetado diretamente no cromatógrafo. Isto só foi possível devido à utilização de um filtro de carbono na linha de gás, posicionado antes da coluna, pois este reduz a entrada, nos instrumentos, de substâncias de baixa volatilidade e de interferentes, levando a um aumento da seletividade do sinal na EM-EM e evitando que houvesse a necessidade de realizar etapas de "clean-up" no procedimento de extração21.
Para detecção foi utilizado um "ion-trap" no modo EM-EM, empregando-se a IE. Quando se utiliza o "ion-trap", a detectabilidade é maximizada pela otimização da quantidade de íons aprisionados no "trap" e dos parâmetros envolvidos na dissociação dos íons precursores com um gás inerte (He) por DIC para obtenção de íons ou clusters de íons com grande abundância relativa no espectro, os quais são empregados na quantificação e identificação dos analitos. Assim, quando se faz a análise de diferentes compostos utilizando EM-EM, vários parâmetros têm que ser otimizados para cada um deles, como amplitude de excitação, nível de excitação para armazenamento ("excitation storage level"), quais íons serão selecionados para quantificação e a partir de qual íon precursor eles serão gerados e as janelas de tempo de retenção ("retention time window") - JTR. Neste trabalho a JRT foi definida como o tempo de retenção médio mais ou menos três vezes a estimativa do desvio padrão dos tempos de retenção de 10 amostras não contaminadas, fortificadas a um nível médio com padrões de calibração de cada composto21.
A identificação dos agrotóxicos foi realizada com base nas JTR e na comparação com espectros de referência, obtidos a partir de um extrato de pepino não contaminado e fortificado com a concentração do segundo ponto da curva analítica. Foram obtidos limites de detecção (LD) e de quantificação (LQ) entre 0,02 e 4 µg kg-1 e entre 0,06 e 13 µg kg-1, respectivamente, para os 81 agrotóxicos. Uma curva analítica foi construída para determinação da linearidade utilizando extratos da matriz livres de contaminação e fortificados com soluções padrão dos agrotóxicos e com a adição de padrão interno, sendo que o primeiro ponto da curva foi determinado a uma concentração entre o LQ e o menor LMR encontrado para os agrotóxicos estudados. A regressão linear foi feita utilizando-se a razão entre a área ou a altura do pico analito/padrão interno versus a concentração. Na validação do método desenvolvido, os autores obtiveram boa linearidade no intervalo de concentrações estudado e também boas precisão e exatidão21.
Este procedimento de análise foi aplicado a amostras reais consumindo-se a metade do tempo gasto em relação a procedimentos já descritos na literatura, o que o torna adequado às análises de rotina21. Na Figura 6 é possível observar o cromatograma obtido a partir de uma amostra real de pimenta contendo 0,05 mg kg-1 de metalaxil, o espectro de massas da amostra e o espectro de referência do metalaxil.
Procedimento semelhante foi aplicado, por este mesmo grupo de pesquisadores19,20, na análise de 71 agrotóxicos de diversas classes (acefato, acrinatrina, amitraz, azoxistrombina, benalaxil, bifentrina, bromopropilato, bupirimato, buprofezina, carbofenotiona, cialotrina, ciflutrina, cipermetrina, clorfenvinfós, cloropirifós, cloropirifós-m, clortalonil, clozolinato, deltametrina, diclorvós, dicofol, difenoconazol, dimetoato, dissulfotom, a-endossulfam, b-endossulfam, endossulfam sulfato, esfenvalerato, etiofencarbe, etion, etoprofós, etrinfós, fenaminfós, fenitrotiona, fenpropatrina, fentiona, flucitrinato, fludioxinil, formotiom, fosalona, furatiocarbe, heptenofós, hexaconazol, iprodiona, isonfenfós, lindano, malationa, metalaxil, nuarimol, oxadixil, parationa-metílica, penconazol, pendimetalina, permetrina, pirazofós, piremetanil, piridabem, pirifenox, pirimicarbe, pirimifós-metílico, piriproxifem, procimidona, propargito, propiconazol, propoxur, quinometionato, tebuconazol, tetraconazol, tetradifona, triadimefom e vinclozolina) em frutas (melão e melancia) e vegetais (ervilha, berinjela, abóbora, pepino, pimenta e tomate), com a diferença de que dois modos de ionização, dependendo do analito, foram empregados em CG-EM-EM (IQ e IE) e foram obtidos resultados semelhantes quanto ao desempenho do método.
A análise de agrotóxicos em matrizes vegetais gordurosas como abacate foi realizada por Moreno et al.22 utilizando CG-EM-EM. Os agrotóxicos estudados neste trabalho foram: acefato, acrinatrina, azoxistrobina, benalaxil, bifentrina, bromopropilato, bupimirato, buprofezina, carbofenotiona, cialotrina, ciflutrina, cipermetrina, clorfenvinfos, cloropirifos, cloropirifos-metílico, clortalonil, clozolinato, deltrametrim, dicofol, diclorvós, difenoconazol, dimetoato, endossulfam I, endossulfam II, endossulfam III, esfenvaralato, etiona, etoprofós, etrinfós, fenpropatrina, fenaminfós, fenitrotiona, fentiona, flucitrinato, fludioxinil, formotiom, fosalona, furatiocarbe, heptenofós, hexaconazol, iprodiona, isonfenfós, lindano, malationa, metalaxil, metamidofós, nuarimol, oxadixil, parationa-metílica, penconazol, permetrina, pirazofós, piridabem, pirifenox, pirimifós-metílico, piriproxifem, procimidona, propargito, propiconazol, propoxur, quinometionato, tebuconazol, tetradifona, triadimefom e vinclozolina.
Nesse trabalho, os autores avaliaram várias técnicas de extração nas quais utilizaram etapas de limpeza, devido ao grande conteúdo lipofílico dos extratos obtidos por extração com solvente orgânico assistida por homogeneizador de alta velocidade ou por extração líquida sob alta pressão e alta temperatura. Eles também avaliaram o uso da cromatografia por permeação em gel ("gel permeation chromatography") - CPG e da extração em fase sólida ("solid phase extraction") - EFS para a limpeza dos extratos22.
Apesar de terem sido obtidas boas recuperações com as duas técnicas de extração, a extração com solvente orgânico assistida por homogeneizador de alta velocidade foi escolhida devido às várias vantagens que ela apresenta, como tempo de extração reduzido, baixo custo, simplicidade e menor manuseio da amostra, o que a torna mais adequada às análises de rotina. Dentre as técnicas de limpeza, a CPG apresentou melhor desempenho, uma vez que com a EFS não foram obtidos valores de precisão e recuperação satisfatórios, além de não eliminar suficientemente impurezas lipofílicas, que causam danos ao sistema cromatográfico, saturação do filtro de carbono, levando à obtenção de sinais analíticos incoerentes22.
Foi utilizado um "ion-trap" operando com IE ou IQ, dependendo do analito, conectado ao sistema cromatográfico. A identificação dos agrotóxicos foi realizada com base nas JTR e comparando os espectros obtidos a partir das amostras analisadas com espectros de referência obtidos sob as mesmas condições cromatográficas. Para quantificação foi utilizada uma curva analítica construída com padrões aplicados ao branco da matriz, na qual o primeiro ponto correspondia à concentração do menor LMR estabelecido. Porém, certos agrotóxicos, como os piretróides, não tiveram boa recuperação quando as amostras não foram processadas por CPG, pois eles possuem elevadas massas molares, de maneira que coeluiram com interferentes lipídicos da matriz. Devido a estes efeitos de matriz, a quantificação dos agrotóxicos foi feita utilizando uma curva analítica que foi construída com o branco da matriz fortificada com os padrões e submetida à etapa de limpeza22.
Outro trabalho de determinação de agrotóxicos em matrizes com alto teor lipídico foi realizado por Patel et al.23, que empregaram CG-EM-EM na determinação de agrotóxicos organoclorados (aldrim, clordano (cis), o,p'-DDT, a-endossulfam, endossulfam-sulfato, a-HCH, b-HCH, g-HCH, heptacloro, heptacloro epóxido (trans), hexaclorobenzeno, oxiclordane e p,p'-TDE) em óleos e gorduras. Para isto as amostras foram limpas por CPG, como no trabalho citado anteriormente22, e analisadas por CG-EM-EM empregando um triplo quadrupolo ajustado ao modo MRM e utilizando IE.
Para evitar a interferência da matriz nos resultados foram selecionados os íons precursores de maior intensidade. Em alguns casos foram selecionados íons de menores intensidades, mas com maiores massas. Fragmentos de íons com m/z menores que 150 foram descartados. Também foi otimizada a energia empregada na cela de colisão para a realização da DIC23.
Nesse trabalho, os autores realizaram a análise de dois agrotóxicos (a-endosulfan e cis-clordano) que coeluiram nas condições de separação utilizadas em CG e que geraram os mesmos íons fragmentos. Mas, foi possível analisar estes agrotóxicos selecionando-se as transições em EM-EM que não foram afetadas pela coeluição destes compostos. Os autores também conseguiram intensificar o sinal dos agrotóxicos que apresentaram elevados valores de sinal/ruído, dividindo o método de aquisição de dados MRM em várias JTR, de acordo com os tempos de retenção dos compostos. Foi estabelecido um número de transições por JTR que garantisse uma detectabilidade adequada às baixas concentrações dos analitos de interesse23.
Os autores também observaram que as respostas obtidas para os padrões dos agrotóxicos em solventes foram menores que as obtidas com padrões aplicados ao branco da matriz. Portanto, os padrões aplicados na matriz foram utilizados para construção da curva analítica, que se apresentou linear no intervalo de interesse (de 0,75 a 30 ng mL-1, com coeficientes de correlação maiores que 0,980) para todos os agrotóxicos avaliados exceto para o o,p'-DDT, o que foi atribuído a uma possível extração incompleta da matriz por CPG. Segundo os autores, a técnica de análise por CG-EM-EM empregada nesse trabalho apresentou excelentes seletividade e LD na identificação e quantificação simultânea dos agrotóxicos organoclorados encontrados em baixos níveis de concentração em gorduras e óleos23.
González-Rodríguez et al.24 determinaram agrotóxicos e alguns metabólitos em diferentes tipos de leite, inclusive materno, utilizando microextração em fase sólida e CG-EM-EM. Os agrotóxicos e metabólitos analisados foram: aldrim, clorfenvinfós, clorpirifós, clorpirifós metílico, clorotanlonil, p,p'-DDD, p,p'DDE, o,p-DDT, p,p'- DDT, diclorana, dieldrim, a-endossulfam, b-endossulfam, endossulfam-éter, endossulfam lactona, endossulfam-sulfato, endrim, endrim aldeído, etiona, fanfur, fenaminfós, heptacloro epóxido, etrinfós, fentiona, heptacloro, hexaclorobenzeno, isofenfós, a-lindano, g,b-lindano, d-lindano, malationa, metoxicloro, mirex, paratiom, paratiom-metílico, pentacloro-benzeno, pentacloronitrobenzeno, pirimifós metílico, quinalfós, sulfotepe e vinclosolina.
Nesse trabalho, os autores ressaltaram a importância de se determinar a presença de agrotóxicos no leite, uma vez que, devido a sua natureza lipofílica, o leite pode ser utilizado para monitorar a bioconcentração de poluentes orgânicos, como esses agrotóxicos organoclorados. Também é importante no sentido de garantir a segurança dos consumidores de produtos lácteos, uma vez que têm sido estabelecidos LMR para agrotóxicos no leite, por órgãos responsáveis no mundo todo24.
A técnica de análise empregada possibilitou a análise de compostos pouco voláteis e sensíveis à temperatura, pois empregou temperaturas de eluição mais baixas (50-80 ºC). As condições de isolamento, fragmentação e estocagem no "ion-trap" foram otimizadas para cada agrotóxico. A identificação dos agrotóxicos foi feita de maneira semelhante à apresentada nos outros trabalhos descritos, por meio das JTR e por comparação com espectros obtidos para os compostos estudados sob as mesmas condições de análise utilizadas. Para evitar interferência da matriz, a curva analítica foi construída com o branco da matriz fortificado com os padrões e usando padrão interno. O método desenvolvido mostrou-se adequado em termos de linearidade, LD e LQ, recuperação e precisão24.
Granby et al.5 desenvolveram um método para análise de carbamatos e outros agrotóxicos relativamente polares (acefato, aldicarbe, aldicarbe-sulfona, aldicarbe-sulfóxido, benfuracarbe, carbaril, carbendazim, etiofencarbe, fenexamida, imazalil, linurom, metamidofós, metiocarbe, metomil, oxamil, pirimetanil, propoxur, tiabendazol e tiofanato-metílico) em diversas matrizes (maçã, abacate, cenoura, alface, laranja, batata e trigo) por CL-EM-EM com IEN. No método de extração empregado, a amostra triturada, misturada com um tampão de acetato metanólico - ácido acético, foi submetida ao ultra-som e, em seguida, à centrifugação. Para compensar as variações na resposta do detector, na quantificação foi utilizado padrão interno marcado isotopicamente ([13C6]-carbaril) e o branco da matriz fortificado.
O espectrômetro de massas utilizado foi um triplo quadrupolo ajustado para que a detecção fosse feita no modo MRM, e para isso, 4 JTR foram selecionadas com 3 a 6 substâncias cada. Quanto mais substâncias são incluídas no método, mais JTR devem ser selecionadas para que se obtenha a detecção com detectabilidade instrumental suficiente. Como foi aplicada a IEN, os autores otimizaram a voltagem a que foi submetida a amostra quando introduzida no espectrômetro ("cone voltage") e, também, a energia de colisão necessária para fragmentar os íons precursores para que fossem obtidos íons produtos com abundâncias relativas significativas5.
Nesse trabalho, os autores verificaram o efeito da matriz comparando-se as respostas obtidas para soluções padrão dos agrotóxicos em solvente (acetato de amônio - ácido acético em metanol - água 95:5 v/v) com soluções padrão dos agrotóxicos no branco da matriz. Como a resposta relativa para a maioria dos agrotóxicos investigados foi por volta de 1, concluiu-se que a matriz não causou supressão ou aumento na resposta obtida no EM-EM. Outro ponto importante desse trabalho foi que não foram observadas diferenças significativas entre as matrizes estudadas, de forma que uma delas pôde ser escolhida para construção das curvas analíticas. Isto é muito importante para que o método seja adequado à aplicação em análises de rotina, nas quais o tempo de análise não deve ser muito longo, para que uma maior quantidade de amostras possa ser analisada5.
Foram obtidas recuperações entre 70 e 120% em três níveis de fortificação (0,02, 0,04 e 0,2 mg kg-1) e LD entre 0,01 e 0,02 mg kg-1 para frutas e vegetais, mas para cereais, 2 dos 19 agrotóxicos avaliados apresentaram LD maiores. A exatidão do método foi avaliada em três testes de proficiência que mostraram uma boa concordância entre os valores obtidos5.
Outro método rápido para detecção e identificação de 38 agrotóxicos (aldicarbe, aldicarbe sulfona, aldicarbe sulfóxido, azoxistrobina, 2,4-D, bendiocarbe, butocarboxim, butocarboxim sulfona, butocarboxim sulfóxido, carbaril, carbendazim, carbofuram, carbofuram 3-hidróxi, cresoxim-metílico, diclofluanida, dietofencarbe, etiofencarbe, fenexamida, 2-fenilfenol, furatiocarbe, imazalil, isoprocarbe, metiocarbe, metiocarbe sulfona, metiocarbe sulfóxido, metomil, metolcarbe, miclobutanil, oxamil, penconazol, propiconazol, pimetrozina, pirimetanil, tebuconazol, tiabendazol, tiodicarbe, tiofanato-metílico e trifloxistrobina) em frutas e vegetais utilizando CL-EM-EM com IEN foi desenvolvido por Taylor et al.17. Este método não utiliza etapas de limpeza após o procedimento de extração da amostra com solvente (neste caso foi utilizado acetato de etila) que, aliado ao uso da eluição isocrática em CLAE (requer um menor tempo para o equilíbrio da coluna após a corrida), torna o método de análise mais rápido. Uma vazão de 0,5 mL min-1 foi utilizada no sistema cromatográfico, sendo necessário o uso de um divisor de fluxo em formato de T para reduzir a vazão da fase móvel do cromatógrafo para aproximadamente 20 µL min-1 antes da introdução no interior do espectrômetro.
A maioria dos agrotóxicos avaliados apresentaram em seus espectros íons pseudo-moleculares ([M+H]+, [M+NH4]+ ou [M+Na]+) no modo de ionização positivo, e aqueles que não foram detectados no modo positivo foram observados no negativo ([M-H]-). Após a aplicação da DIC, utilizando argônio, foram identificados estruturalmente íons produtos para todos os compostos, com exceção de apenas um agrotóxico (diclofluanida), de forma que puderam ser monitorados utilizando MRM17.
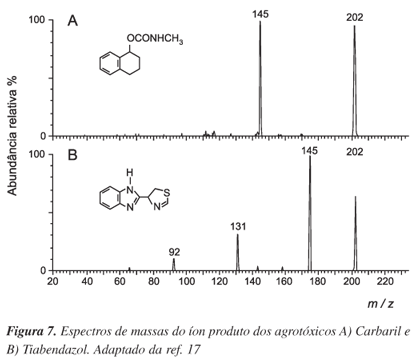
Uma característica particular da detecção por EM-EM que foi explorada nesse estudo foi a capacidade de diferenciar entre analitos isobáricos, especialmente os que coeluem. As diferenças entre o espectro de massas do íon produto gerado por íons precursores comuns foram decisivas na determinação dos seguintes pares de agrotóxicos estudados: aldicarbe e butocarboxim; aldicarbe sulfóxido e butocarboxim sulfóxido e, carbaril e tiabendazol17. Os espectros de massas do íon produto do carbaril e tiabendazol são mostrados na Figura 7. O carbaril é caracterizado pela perda de CH3NCO, formando um íon de m/z 145 e o tiabendazol pela perda de HCN, resultando em um íon de m/z 175 a partir de um íon precursor comum de m/z 20217.
Sannino et al.25 desenvolveram um método para determinação de 24 agrotóxicos (azoxistrobina, benfuracarbe, bromuconazol, cresoxim-metílico, difenoconazol, dimetomorfe, etofemprox, fenbunconazol, fenazaquim, fenexamida, fenotiocarbe, fenpiroximato, flusilazol, furatiocarbe, hezitiazoxi, imidacloprido, indoxacarbe, paclobutrazol, piridabem, tebuconazol, tetraconazol, trifloxistrobina, tebufenozida e tebufenpirade) em purê de maçã, tomate e em suco concentrado de limão. Este método extrai os agrotóxicos de interesse da amostra utilizando apenas uma mistura de solventes (acetato de etila - cicloexano 50:50 v/v) e filtrando a amostra para evitar a entrada de material particulado nos instrumentos.
Para identificação e quantificação dos agrotóxicos foi utilizado um sistema CLAE acoplado a um triplo quadrupolo equipado com uma interface IEN. Simultaneamente, 55 transições foram monitoradas por EM-EM durante a separação por cromatografia líquida, no modo de varredura MRM, utilizando apenas uma JTR, após a otimização das condições de operação instrumentais realizada pela injeção de soluções individuais de cada agrotóxico em metanol25.
Nesse trabalho, também foi estudado o efeito da matriz sobre o sinal detectado, comparando-se o sinal obtido para os padrões de agrotóxicos em solvente e no branco da matriz, não sendo observado efeito de supressão significativo no sinal, exceto para o etofenproxi. Um aumento no sinal foi observado para dois dos agrotóxicos avaliados, fenpiroximato e indoxacarbe. Os autores relataram que a melhor forma para compensar os efeitos da matriz seria utilizar padrões internos marcados isotopicamente, porém, para muitos agrotóxicos não existe este tipo de composto disponível. Mesmo assim, com o uso de padronização externa, empregando o branco da matriz fortificado, foi possível analisar 102 amostras reais de frutas processadas, nas quais não foram encontrados resíduos de agrotóxicos acima de 10 µg kg-1 25.
Ferrer et al.26 determinaram resíduos de agrotóxicos (cipermetrina, deltametrina, endossulfam I, endossulfam II, endossulfam sulfato, paratiom-metílico, pirimifós-metílico, simazina e terbutilazina) em azeitonas e azeite por dispersão da matriz em fase sólida, com uma etapa de limpeza realizada pela eluição do extrato através de uma coluna de Florisil, que foi, em seguida, submetido à análise por CL-EM-EM. Os autores ressaltaram a importância de se determinar agrotóxicos em azeite uma vez que sua obtenção é realizada apenas por processos físicos e/ou mecânicos, sem nenhuma etapa posterior de tratamento ou purificação, de maneira que é muito provável a incidência de resíduos de agrotóxicos nesta matriz.
Para realização da CL-EM-EM foi utilizada uma coluna de fase reversa C8, com eluição por gradiente e um "ion-trap" operando no modo positivo com IEN. Os autores enfatizaram que quando se utilizou apenas CL-EM foram detectados sinais de interferentes isobáricos da matriz nos espectros de massas, daí a necessidade de se empregar EM-EM. Isto possibilitou isolar o íon precursor e otimizar a fragmentação, gerando um aumento na detectabilidade e na seletividade, assim como menores razões sinal/ruído. Mesmo assim, para o agrotóxico dimetoato houve supressão do sinal por interferentes da matriz e sua análise só foi possível utilizando espectrometria de massas em multi estágios (EMn), pois EMn possibilitou a remoção total da interferência da matriz, mas foi acompanhada de um aumento na razão sinal/ruído gerando, conseqüentemente, maior LD 26.
APLICAÇÃO DA CL-EM-EM À DETERMINAÇÃO DE MEDICAMENTOS VETERINÁRIOS EM ALIMENTOS
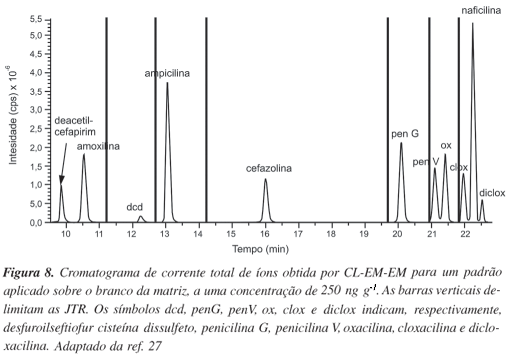
Fagerquist et al.27 realizaram a determinação quantitativa e qualitativa de antibióticos (em específico as b-lactamas: amoxilina, ampicilina, cefazolina, cloxacilina (clox), deacetil-cefapirim, desfuroilseftiofur cisteína dissulfeto (dcd), dicloxacilina (diclox), naficilina, oxacilina (ox) e penicilina G (pen G)) em carne bovina proveniente do Serviço de Segurança e Inspeção de Alimentos dos Estados Unidos da América (EUA), utilizando a extração em fase sólida e CL-EM-EM. No sistema cromatográfico foi utilizada uma vazão de 0,3 mL min-1 para gerar uma linha de base estável e uma ionização eficiente dos analitos por IEN. A análise por EM-EM foi realizada utilizando um triplo quadrupolo ajustado ao modo de varredura MRM, para monitorar as 4 transições mais abundantes do íon precursor - íon produto de cada analito. A Figura 8 mostra um cromatograma típico da corrente total de íons, que representa a soma dos sinais gerados por todas as transições do íon precursor - íon fragmento obtidas em cada JTR.
Nesse trabalho os autores observaram que a naficilina gerou saturação do detector em sua transição mais abundante, devido à alta eficiência de ionização apresentada por este composto. Isso foi observado devido a uma não linearidade pronunciada na sua curva analítica. Segundo os autores, este problema poderia ser facilmente resolvido diminuindo-se a voltagem através do detector. Porém, para um método de determinação multirresidual com detecção por EM, o instrumento tem que ter os seus parâmetros ajustados de forma adequada para monitorar todos os analitos, mas não necessariamente na condição ótima para cada analito. Assim, a diminuição da voltagem através do detector poderia resultar na perda de detectabilidade para as transições menos abundantes dos analitos com menor eficiência de ionização, de maneira que foi concluído que as curvas analíticas não lineares poderiam ser usadas para a determinação da concentração, uma vez que a interpolação das curvas não lineares era suficientemente exata27.
O método desenvolvido foi comparado ao ensaio microbiológico utilizado pelo Serviço de Segurança e Inspeção de Alimentos dos EUA para determinação destas drogas veterinárias em carne. O método utilizando CL-EM-EM foi capaz de detectar antibióticos em níveis de concentração mais baixos que o ensaio microbiológico e também foi capaz de determinar a identidade de cada antibiótico presente, o que não foi possível no ensaio microbiológico. Com este método, também foi possível determinar quantitativamente os antibióticos abaixo dos níveis de tolerância estabelecidos por lei27.
Outro método de determinação de b-lactamas (amoxilina e ampicilina) em leite e carne bovina foi descrito por Bogialli et al.28, no qual foi adicionado às amostras um padrão interno (penicilina V) e, posteriormente, realizada a extração por dispersão da matriz em fase sólida com eluição utilizando água aquecida. Depois de ajustado o pH do extrato, este foi filtrado e analisado por CL-EM-EM utilizando coluna de fase reversa C18 no sistema cromatográfico, IEN e varredura MSR. O espectro MSR obtido por CL-EM-EM para o leite fortificado com 4 µg kg-1 com as b-lactamas estudadas encontra-se na Figura 9.
Os autores observaram um entupimento parcial da coluna de guarda C18 utilizada no sistema cromatográfico, o que foi atribuído à presença de sais insolúveis em água nos produtos de extração do fígado e dos rins bovinos. Mesmo assim o método apresentou boa precisão, exatidão e LD < 1 µg kg-1 para ambos os analitos nas matrizes avaliadas28.
Matabudul et al.29 desenvolveram um método para a determinação simultânea de antibióticos ionóforos (lasalocide, monensina, narasina e solinomina) em fígado de animais de diferentes espécies e em leite, previamente extraídos com acetonitrila e por EFS utilizando cartuchos de sílica e analisados por CL-EM-EM utilizando IEN no modo positivo e varredura MRM. Os antibióticos ionóforos complexam com cátions (K+, Na+, Ca2+ e Mg2+) e são lipossolúveis.
A voltagem da IEN para obtenção de íons produto [M+Na]+ foi ajustada utilizando soluções de padrões dos agrotóxicos dissolvidos na FM (acetonitrila, metanol, tetraidrofurano, água e ácido trifluoracético 67:10:10:13:0,1 v/v). Não foram observados interferentes nos espectros e recuperações semelhantes foram obtidas para as diferentes matrizes, de forma que foi possível considerar o método desenvolvido robusto e versátil. Como o tempo de preparo das amostras neste método foi menor que 3 h, ele pode ser empregado na análise de mais de 40 amostras por dia29.
Andersen et al.30 desenvolveram um método para determinação de algumas tetraciclinas, antibióticos veterinários também utilizados em aquacultura, em amostras de camarão e leite integral usando CLAE, com detecção por ultravioleta e a confirmação do resíduo por EM e EM-EM. As amostras de leite e camarão foram extraídas adicionando-se ácido succínico e, em seguida, foi realizada uma etapa de limpeza por EFS. O espectrômetro de massas, um triplo quadrupolo usando IEN no modo positivo, foi ajustado para realizar a varredura no modo MSR, de maneira que foi possível confirmar a presença do resíduo de três tetraciclinas (tetraciclina, oxitetracilina e clorotetraciclina) em concentrações entre 25 e 400 ng g-1.
Nicolich et al.31 desenvolveram um método para determinação de cloranfenicol em leite por LC-EM-EM no modo MRM, com IEN e utilizando padrão interno marcado isotopicamente (deuterado - D5). O cloranfenicol é um antibiótico de largo espectro que é capaz de causar doenças sanguíneas fatais em seres humanos. Além disso, não é possível estabelecer um limite para a presença de seus resíduos e de seus metabólitos nos alimentos que garantam a segurança dos consumidores, ou seja, o LMR do cloranfenicol não pode ser determinado. Desta maneira, o uso do cloranfenicol em animais geradores de alimentos é proibido em diversos países, inclusive no Brasil.
As amostras de leite foram fortificadas com o padrão interno e submetidas à extração líquido-líquido com acetato de etila e uma solução de ácido fórmico (10 mmol L-1). A separação cromatográfica foi realizada empregando-se uma coluna cromatográfica Varian Pursuit em combinação com uma coluna de guarda Varian MetaGuard e, como FM, foram utilizadas soluções de 0,1% de ácido fórmico em água e 0,1% de ácido fórmico em acetonitrila. A curva analítica foi obtida utilizando-se o branco da matriz fortificado com soluções de padrão de cloranfenicol. O método desenvolvido apresentou limite de decisão de 0,05 ng mL-1 e capacidade de detecção de 0,09 ng mL-1, os quais foram calculados a partir das curvas analíticas, assim como boa exatidão e precisão, de forma que este método pode ser aplicado no monitoramento do cloranfenicol em amostras de leite obtidas em supermercados brasileiros31.
Mottier et al.32 realizaram a determinação quantitativa de quatro metabólitos de nitrofurano (furaltadona, furazolidona, nitrofurantoína e nitrofurazona) em carne por CL-EM-EM com IEN, utilizando padrões internos marcados isotopicamente. Os nitrofuranos são utilizados como aditivos em ração animal para o tratamento profilático e terapêutico de doenças causadas por bactérias e protozoários e alguns deles foram proibidos na Europa por serem mutagênicos e carcinogênicos. As amostras foram primeiramente extraídas com acetato de etila e, em seguida, foram submetidas a uma etapa de limpeza por EFS para posterior injeção no sistema CLAE, no qual foi utilizado gradiente linear para eluição dos analitos em fase reversa.
Para quantificação dos analitos foi empregada a padronização externa utilizando padrões dos agrotóxicos em água e no branco da matriz com adição de padrões internos dos analitos marcados isotopicamente. Alguns dos padrões internos marcados isotopicamente (deuterados) não se comportaram da mesma forma que os compostos endógenos durante o processo de análise e por isso, não foram considerados apropriados32.
Não foram observados picos de interferentes nas transições monitoradas quando se utilizou o branco da matriz contendo apenas os analitos e os padrões internos. Quando se compararam as curvas analíticas com os padrões dos agrotóxicos em água e no branco da matriz, foi verificado que não houve uma completa compensação dos efeitos da matriz pelo uso dos padrões internos. Porém, como os resultados obtidos para ambas as curvas analíticas não foram significativamente diferentes, utilizou-se a curva analítica empregando padrões em água para a validação do método. O método proposto foi considerado seletivo e confirmatório para detecção dos analitos em níveis abaixo dos LMR32.
Heller et al.33 realizaram a quantificação de resíduos de sulfonamidas (sulfacetamida, sulfacloropiridazina, sulfadiazina, sulfadimetoxalina, sulfadimetoxina, sulfagüanidina, sulfamerazina, sulfametazina, sulfametiazol, sulfametoxazol, sulfametoxipiridazina, sulfamonometoxina, sulfanilamida, sulfapiridina, sulfatiazol e sulfisoxazol) em ovos por CLAE com detecção ultravioleta e confirmação por EM-EM.
As amostras foram extraídas com acetonitrila, seguida de uma etapa de limpeza por EFS utilizando um cartucho de C18. Os analitos foram separados por CLAE em fase reversa (coluna C18) utilizando eluição por gradiente, ionização por IEN e detecção por EM-EM em um espectrômetro de massas "ion-trap". Os íons moleculares protonados foram isolados e dissociados para obtenção do espectro de massas a partir da varredura de todos os íons produtos33.
Duas das sulfonamidas analisadas nesse trabalho (sulfametoxipiridazina e sulfamonometoxina) possuem a mesma massa molar e geraram espectros que continham os mesmos íons produtos e por isso não puderam ser diferenciadas uma da outra33.
A análise de quatro 5-nitroimidazóis (dimetridazol, ipronidazol, metronidazol e ronidazol) e seus correspondentes metabólitos hidroxilados em ovos, ovos processados e carne de frango foi realizada por Mottier et al.34 utilizando CL-EM-EM e padrões internos marcados isotopicamente. Os 5-nitromidazóis são antibióticos de uso veterinário suspeitos de serem carcinogênicos e mutagênicos em seres humanos. Estes compostos são metabolizados rapidamente e alguns de seus metabólitos são carcinogênicos e mutagênicos em certas espécies de animais, daí a importância de se monitorar não só os antibióticos, mas também seus metabólitos.
As amostras de ovos foram fortificadas com padrões internos dos analitos deuterados e depois submetidas à extração com acetonitrila e posterior etapa de limpeza por EFS. As amostras de carne foram também fortificadas com os padrões internos deuterados, mas foram extraídas com acetato de etila e apenas filtradas após a extração, antes de serem injetadas no cromatógrafo. No sistema cromatográfico foi utilizada a eluição por gradiente linear em fase reversa para separar os analitos. O espectrômetro de massas, um triplo quadrupolo, foi ajustado ao modo MRM para o monitoramento de duas transições para cada 5-nitroimidazol e seus correspondentes padrões internos. Os analitos foram ionizados por IEN no modo positivo34.
O método de extração utilizado permitiu que mais de 150 injeções de amostras fossem realizadas sem perdas no desempenho da coluna cromatográfica ou contaminação excessiva do espectrômetro de massas. Como não foram observadas diferenças significativas entre as curvas analíticas obtidas com padrões dos analitos em água e no branco da matriz, a calibração com padrões aquosos foi utilizada, permitindo que a quantificação de 5 dos 7 analitos avaliados fosse realizada de maneira precisa e exata. Estes 5 compostos possuíam um padrão interno análogo, isotopicamente marcado, os demais não34.
APLICAÇÃO DE CL-EM E CG-EM-EM NA DETERMINAÇÃO DE TOXINAS EM ALIMENTOS
Chen et al.35 desenvolveram um método para determinação de aflatoxinas em leite líquido e em pó, utilizando EFS com alta vazão e CL-EM-EM. As aflatoxinas são metabólitos secundários da Aspergillus flavus e Aspergillus parasiticus, sendo encontradas freqüentemente em cereais contaminados e frutas secas. A aflatoxina B1 (AFB1) é a que possui maior toxicidade, uma vez que pode causar câncer de fígado quando um indivíduo é exposto por longo tempo a esta toxina, principalmente quando se tratam de pessoas com hepatite B. O principal metabólito da AFB1 é a aflatoxina M1 (AFM1), que é menos carcinogênica que AFB1 e pode ser encontrada na urina, sangue, leite e órgãos internos de animais infectados.
Os autores utilizaram um triplo quadrupolo com IEN no modo MSR. Como em IEN o pH da FM é crítico para a intensidade do sinal, foram testadas soluções aquosas de ácido fórmico (pH 2,9) e 4-metilmorfolina (pH 9,7) para os modos negativo e positivo, respectivamente, com a injeção de 30 ng de AFM1 no sistema cromatográfico. Os picos produzidos a partir dos íons negativos foram melhores que do íon protonado, tanto em relação à forma, quanto à proporção sinal/ruído. Observaram que os diferentes materiais utilizados na etapa de limpeza por EFS (colunas de imunoafinidade Afla M1 - IAC e Mycosep 226 - MFC) tiveram influência sobre os efeitos da matriz no sinal, uma vez que quando se utilizou a coluna IAC não foram observados efeitos de matriz, mas utilizando a coluna MFC foi observada a supressão do sinal em amostras reais. De acordo com os resultados obtidos, os autores afirmaram não haver a necessidade de utilizar o branco da matriz para construção da curva analítica, uma vez que o método foi linear35.
A utilização da detecção por EM-EM possibilitou a obtenção de LD até 85 vezes menores que os limites reguladores da União Européia (50 ng L-1) para o leite líquido e até 3 vezes mais sensíveis que o nível máximo estabelecido para bebês (25 ng kg-1) no leite em pó, determinado pela União Européia35.
O desenvolvimento de uma metodologia para determinação de azaspiracide em mariscos por CL-EM-EM foi realizado por Draisci et al.36. A azaspiracide é uma toxina responsável por casos de intoxicação de seres humanos resultante do consumo de mariscos na Europa. Este analito foi extraído de mexilhão utilizando acetona e, a seguir, foi submetido à análise cromatográfica usando uma microcoluna C18 (fase reversa) e eluição isocrática, a uma vazão de 30 µL min-1, com acetonitrila:água (85:15 v/v) contendo 0,03% de ácido trifluoracético. A toxina foi ionizada numa interface IEN operando no modo positivo, sendo gerada somente, como íon precursor, a molécula protonada. Por DIC, em um triplo quadruplo, o íon precursor foi dissociado em três íons produto, [M+H-nH2O]+ com n = 1-3, os quais foram identificados utilizando MSR. Com o método desenvolvido foi possível obter boa linearidade no intervalo de concentração entre 0,1 e 1 µg mL-1 (padronização externa) e LQ instrumental consideravelmente menor que o limite obtido em ensaios biológicos.
Rundberget e Wilkins37 realizaram a determinação de micotoxinas do Penicillium (ácido micofenolítico, chaetoglobosina B, griseofulvina, penitrem A, roquefortine C e verruculogena) em alimentos utilizando LC-MS-MS. Os autores ressaltaram que, devido à grande toxicidade das micotoxinas do Penicillinum, há a necessidade do uso de técnicas de análise com as quais se consiga atingir baixos limites de detecção. Para algumas destas toxinas, na União Européia, é legalmente aceitável concentrações de 5 ng g-1 em grãos. Os alimentos selecionados para realização da análise foram extraídos primeiramente com uma mistura de acetonitrila/água e, em seguida, com hexano para eliminar a gordura. O extrato aquoso filtrado foi injetado no sistema cromatográfico, no qual foi empregada uma coluna C18 (fase reversa) e eluição por gradiente iniciada com uma mistura de metanol e água na proporção de 40:60 v/v e terminada, após 15 min, com a proporção de 95:5 v/v da mesma mistura de solventes. O sistema cromatográfico encontrava-se acoplado a um espectrômetro "ion-trap" operando com interface IEN ou IQPA, no qual foi utilizado o modo MSR para obtenção do espectro de massas.
Nesse trabalho, os autores observaram que a altura e a área dos picos obtidos para as micotoxinas por CL-EM-EM dependem da matriz, pois numa mistura das matrizes analisadas elas foram até 40% menores que as obtidas com padrões das micotoxinas em metanol:água. Ficou, portanto, evidente o efeito da supressão dos componentes das matrizes sobre a ionização das diferentes espécies de micotoxinas analisadas. Por esse motivo, os autores realizaram a quantificação utilizando o branco da matriz e padrão interno. O método desenvolvido foi capaz de determinar as micotoxinas de interesse em concentrações abaixo de 5 ng g-1, como desejado37.
Lau et al.38 desenvolveram um procedimento para a determinação de micotoxinas da Alternaria (alternariol e monometil éter alternariol) em sucos de frutas e bebidas alcoólicas por CLAE-UV, CL-EM e CL-EM-EM. Estas micotoxinas são apontadas como possíveis causadoras de câncer no esôfago de seres humanos.
As amostras foram submetidas à extração em fase sólida, na qual foram empregados cartuchos de C18 e aminopropil em série. Os eluatos das bebidas alcoólicas foram evaporados em um rota-evaporador para eliminação do etanol. No sistema CL-EM o modo de varredura utilizado foi o MIS e no sistema CL-EM-EM foi empregado um triplo quadrupolo com ionização por IQPA ou IEN, no modo negativo e varredura MRM38.
Os autores observaram que os sinais obtidos na análise por CLAE-UV foram mais intensos que os obtidos por CL-EM e CL-EM-EM, o que mostra que o sinal em CLAE-UV sofre uma maior interferência da matriz. Em CL-EM, não foi possível detectar os analitos presentes em baixos níveis de concentração em certas matrizes, devido a uma elevada interferência da matriz. Mas em CL-EM-EM, a alta detectabilidade obtida permitiu a determinação de sub ng mL-1 das micotoxinas analisadas38.
Outro trabalho de determinação de micotoxinas em alimentos foi realizado por Häubl et al.39, que utilizaram CL-EM-EM para determinação de dioxinivalenol em milho, empregando um padrão interno marcado isotopicamente (13C).
Após a adição de padrão interno à amostra, esta foi submetida à extração com uma mistura de acetonitrila:água (84:16 v/v) para posterior filtração, evaporação e redissolução do extrato em água. O produto da extração foi injetado em um sistema cromatográfico operando em fase reversa com eluição por gradiente linear, empregando uma mistura de metanol:água como fase móvel. O sistema cromatográfico foi acoplado a um "ion-trap" com ionização por IEN no modo negativo e varredura MRM39.
Foram utilizadas soluções padrão dos agrotóxicos em água contendo padrão interno e foram construídas duas curvas analíticas: uma com o padrão interno e outra por padronização externa. Utilizou-se um material de referência contendo (470 ± 30) µg kg-1 da micotoxina, para avaliar as duas curvas analíticas. Para a curva construída considerando-se os padrões internos obteve-se uma concentração de (463 ± 16) µg kg-1 da micotoxina com recuperação de 99% para n=7 e para a curva com padronização externa obteve-se uma concentração de (176 ± 22) µg kg-1 da micotoxina com recuperação de 37% para n=7. Esta diferença significativa observada nos resultados mostra que o uso de padrão interno é capaz de compensar variações experimentais e instrumentais, mas compensa principalmente a supressão iônica ocasionada pela matriz que ocorre em CL-EM-EM, apesar da grande seletividade e detectabilidade desta técnica39.
Eppe et al.40, numa tentativa de aumentar a detectabilidade em um sistema CG-EM-EM, utilizaram um injetor no sistema cromatográfico que possibilitava a injeção de grandes volumes de amostra, para analisar dioxinas do gênero policlorodibenzo-p-dioxina/furano (PCDD/F) em alimentos. Os resultados foram comparados aos obtidos para as mesmas amostras analisadas por CG com detecção por um espectrômetro de massas de alta resolução.
As amostras de alimentos analisadas, carne bovina gordurosa, gema de ovo e leite em pó, foram submetidas à extração em fase líquida sob alta pressão com uma mistura de diclorometano:pentano (1:1 v/v), passando em seguida por uma etapa de limpeza em um sistema no qual foi empregada uma coluna de sílica ácida de alta capacidade, uma pequena coluna com múltiplas camadas de sílica, uma coluna de alumina alcalina e uma coluna de carbono PX-21. No sistema CG-EM-EM, foi utilizado um "ion-trap" com ionização por IE, empregando gás hélio para promover a dissociação dos analitos por DIC. Os resultados obtidos mostraram uma boa concordância entre as técnicas utilizadas, de maneira que a CG-EM-EM com injeção de maiores volumes pode ser atrativa para detecção de µg kg-1 de toxinas em alimentos devido ao seu menor custo em comparação à técnica que utiliza o espectrômetro de massas de alta resolução40.
APLICAÇÃO DE CG-EM-EM E CL-EM-EM À DETERMINAÇÃO DE SUBSTÂNCIAS TÓXICAS GERADAS DURANTE O PREPARO DOS ALIMENTOS
Hoenicke et al.41 realizaram a análise de acrilamida em diferentes tipos de alimentos utilizando CL-EM-EM e CG-EM-EM. Atribuí-se a presença de acrilamida em alimentos à reação de Maillard, que é responsável pelos sabores e coloração "marrom" característicos de alimentos fritos e assados. Segundo os autores, o monitoramento da presença de acrilamida em alimentos se faz necessária por ser considerada um composto potencialmente carcinogênico. Nesse trabalho foram utilizadas duas técnicas de extração, sendo que uma delas foi realizada com iso-hexano, seguida por uma etapa de purificação com hexacianoferrato de potássio e sulfato de zinco (precipitação de Carrez). Na outra técnica, a extração foi feita com água em banho de ultra-som, seguida de purificação empregando a precipitação de Carrez e uma etapa de extração com acetato de etila. Segundo os autores, o uso da precipitação de Carrez para purificação da amostra permitiu que mais de 60 amostras fossem preparadas por dia. Porém, esta etapa foi insuficiente para purificar amostras de matrizes muito complexas, como café solúvel e cacau, daí a necessidade de se realizar uma etapa "extra" de extração. Esta etapa adicional foi capaz de eliminar as interferências espectrais.
A análise por CL-EM-EM foi realizada utilizando IEN no modo positivo e um triplo quadrupolo ajustado para obtenção dos espectros por MRM. No sistema cromatográfico foi empregada eluição por gradiente utilizando como fase móvel uma mistura de acetonitrila:ácido acético 1% em fase reversa (coluna LiChrospher 100 CN), numa vazão de 0,7 mL min-1. No sistema CG-EM-EM o cromatógrafo a gás foi acoplado a um triplo quadrupolo, sendo utilizada a IQ para ionização do analito no modo positivo e MSR para obtenção dos espectros de massas. Com CL-EM-EM foi possível obter um LD de 10 µg kg-1 e com CG-EM-EM, o LD foi de 5 µg kg-1 41.
Outro trabalho visando a determinação de acrilamida em alimentos por CL-EM-EM foi realizado por Bermudo et al.42. Para isso foi empregado um espectrômetro "ion-trap" e a ionização do analito foi realizada no modo positivo por IQPA. No sistema cromatográfico foi utilizada fase reversa e uma FM com 100% de água, a uma vazão de 0,3 mL min-1. Antes da etapa de extração foi adicionado às amostras um padrão interno de acrilamida marcada isotopicamente para posterior quantificação. A extração foi feita com água, por agitação e, em seguida, a limpeza foi realizada por EFS.
O método desenvolvido por Bermudo et al.42 foi aplicado a 30 amostras de alimentos coletadas em supermercados de Barcelona e quando a acrilamida estava presente nas amostras em concentrações inferiores ao LD de 45 ng g-1, obtido utilizando o "ion-trap", empregava-se um triplo quadrupolo ajustado ao modo de varredura MRM para efetuar a determinação. Os maiores teores de acrilamida foram encontrados em batatas fritas (500 a 9250 ng g-1).
Zhang et al.43 determinaram acrilamida em alimentos infantis à base de cereais por CL-EM-EM, empregando, para isto, um triplo quadrupolo e ionização por IEN no modo positivo. A gordura presente nas matrizes analisadas foi extraída com éter de petróleo e, em seguida, a extração foi realizada com uma solução aquosa de cloreto de sódio (2 mol L-1) e acetato de etila. A etapa de limpeza do extrato foi realizada por EFS. Antes de serem submetidas à extração, as amostras receberam a adição de um padrão interno de acrilamida marcada isotopicamente, com o objetivo de verificar a ocorrência de perdas durante o pré-tratamento da amostra e também para seu emprego na quantificação, uma vez que as curvas analíticas foram obtidas pela razão entre a área do pico cromatográfico da acrilamida não marcada e a marcada presente nos padrões utilizados. Após a validação, os autores concluíram que o método desenvolvido é seletivo, preciso e robusto e pode ser empregado na determinação de acrilamida em baixos níveis de concentração em alimentos (LQ de 3 µg kg-1).
Andrzejwski et al.44 otimizaram um método para determinação de acrilamida em café solúvel, pronto e em grãos por CL-EM-EM. Foi adicionado padrão interno marcado isotopicamente (13C) às amostras para sua posterior extração com água, seguida de uma etapa de limpeza por EFS, com colunas Oasis HBL. A análise por CL-EM-EM foi realizada em fase reversa no sistema cromatográfico, empregando-se uma coluna Synergi Hydro-RP, com partículas de 4 µm. Foi usada eluição isocrática, com fase móvel de 0,5% de metanol em água, acoplado a um triplo quadrupolo com ionização por IEN e varredura MRM.
Graças ao emprego de um padrão interno estável, os autores puderam observar a perda do sinal da acrilamida após sucessivas injeções do extrato de café. Este problema pôde ser solucionado ajustando-se a temperatura da coluna. Foi também observada a presença de interferentes co-extraídos nos cromatogramas obtidos, porém, estes sinais não interferiram no sinal da acrilamida ou do padrão interno graças à boa resolução cromatográfica obtida. O método desenvolvido apresentou boa linearidade e possibilitou verificar a presença de acrilamida em diversas marcas populares de café43.
CONCLUSÕES
Considerando as aplicações da cromatografia com detecção por espectrometria de massas acoplada à espectrometria de massas apresentadas nesta revisão, certifica-se que esta técnica mostrou-se bastante eficiente na análise de substâncias tóxicas, em nível de traços em alimentos, no sentido de terem sido atingidos, na maioria das técnicas de análise desenvolvidas, LQ abaixo dos LMR dos compostos de interesse, de ter sido possível detectar a maioria dos compostos que se desejou analisar, de ter sido detectado um grande número de analitos de diferentes classes simultaneamente, no caso de análises multirresiduais, e da possibilidade de confirmar a identidade dos compostos analisados com exatidão, graças aos diferentes modos de varredura e ionização que podem ser utilizados.
Em alguns dos trabalhos descritos, ficou evidente a dificuldade em obter o sinal do analito livre de interferências geradas pela matriz analisada. Apesar da grande seletividade da técnica EM-EM, alimentos são matrizes muito complexas, das quais nem sempre é possível extrair o analito sem que interferentes estejam presentes nos extratos obtidos. Este problema mostrou-se mais grave quando os analitos de interesse estão presentes em matrizes com grande teor de gordura, como carnes, leite e óleos, para as quais foi necessário empregar etapas de limpeza por EFS ou CPG no processo de preparo da amostra. Entretanto, muitas das técnicas de extração utilizadas são trabalhosas e demoradas, de forma que a sua aplicação em análises de rotina se torna difícil.
Outra limitação da aplicação da cromatografia com detecção por espectrometria de massas acoplada à espectrometria de massas em análises de rotina é que vários parâmetros instrumentais têm que ser otimizados para cada analito, como energia de ionização, ajuste das JTR, energia de dissociação (quando se utiliza DIC), para que sejam obtidos espectros de massas com abundâncias relativas dos íons de interesse adequadas. Porém, para um método de determinação multirresidual com detecção por EM, o instrumento tem que ter seus parâmetros ajustados de forma adequada para monitorar todos os analitos simultaneamente, mas não necessariamente na condição ótima para cada analito. Isto pode levar a uma detecção pouco sensível de alguns compostos, principalmente quando pertencem a classes diferentes.
A capacidade desta técnica de fornecer informação estrutural com exatidão permite que se realize a distinção entre compostos isobáricos de interesse ou provenientes da matriz, quando estes produzem um espectro de massas com íons fragmentos distintos. Mas, alguns compostos isobáricos geram espectros de massas com íons fragmentos de mesma massa. Neste caso, EM-EM não é capaz de distinguí-los. Em alguns casos, a aplicação de EMn possibilita diferenciar entre estes tipos de analitos, porém, na maioria dos casos causa uma diminuição da detectabilidade, seletividade e aumento na razão sinal/ruído.
A maior seletividade de EM-EM reduz a necessidade de se obterem separações cromatográficas com grande resolução, uma vez que esta técnica permite identificar e, muitas vezes quantificar, compostos que coeluem.
Os dados obtidos com EM-EM não são mais complexos que aqueles provenientes da EM "convencional", de forma que os analistas que dominam a técnica de EM podem obter resultados com maior seletividade utilizando EM-EM. Espectrômetros como o "ion-trap" apresentam a versatilidade de poderem ser ajustados para operar nos dois modos, EM e EM-EM, dispensando maiores investimentos na parte instrumental para obtenção de maior seletividade.
Tanto espectrômetros "ion-trap" quanto triplos quadrupolos são, com maior freqüência, empregados na análise de compostos tóxicos em alimentos. Porém, os triplos quadrupolos permitem a obtenção de menores LD, de forma que podem ser mais adequados em casos de compostos com menores LMR.
Uma das principais vantagens de se acoplar a cromatogrefia à EM-EM é que a EM-EM permite a identificação de compostos com alto grau de confiança, o que não seria possível apenas com base nas características de retenção/eluição dos compostos, fornecidas por outros detectores utilizados em CLAE e CG. Outra vantagem muito importante é que, quando os analitos de interesse fazem parte de uma mistura, apenas utilizando a técnica espectrométrica, o espectro de massas obtido apresentaria íons de todos os componentes desta mistura e a identificação dos analitos seria feita com maior dificuldade. Mas, graças à combinação da espectrometria com a capacidade de separação da cromatografia, na maioria das vezes compostos "puros" são introduzidos no espectrômetro de massas, de maneira que podem ser identificados de forma inequívoca.
O alto custo do equipamento empregado em EM-EM torna sua utilização inviável em muitos laboratórios. Porém, hoje, seu valor já se encontra menor devido à concorrência de mercado e muitas das vantagens associadas ao uso da EM-EM podem compensar o alto investimento inicial.
SÍMBOLOS
afinidade eletrônic
afinidade protônica
analisador de tempo de vôo ("Time-of-Flight Analiser")
cromatografia gasosa com detecção por espectrometria de massas acoplada à espectrometria de massas
cromatografia de íons totais ("total ion chromato-gram")
cromatografia líquida de alta eficiência ("high performance liquid chromatography")
cromatografia líquida de alta eficiência com detecção por ultra-violeta
cromatografia líquida acoplada à espectrometria de massas
cromatografia líquida com detecção por espectrometria de massas acoplada à espectrometria de massas
cromatografia por permeação em gel ("gel permeation chromatography")
dissociação induzida por colisão com um gás inerte ("collision-induced dissociation")
primeiro estágio de espectrometria de massas na espectrometria de massas acoplada à espectrometria de massas
segundo estágio de espectrometria de massas na espectrometria de massas acoplada à espectrometria de massas
espectrometria de massas acoplada à espectrometria de massas
fotoionização em pressão atmosférica ("atmospheric pressure photoionization")
ionização por eletronebuilização ("electrospray ionization")
ionização química à pressão atmosférica ("atmospheric pressure chemical ionization")
janela de tempo de retenção ("retention time window")
monitoramento de íons selecionados ("selected ion monitoring")
níveis máximos de resíduo ("maximum residue levels")
limite de quantificação
monitoramento dos íons selecionados ("selected ion monitoring")
monitoramento de reações múltiplas ("multiple-reaction monitoring")
monitoramento seletivo de reações
("selected-reaction monitoring")
instrumento em que o último estágio de um triplo quadrupolo é substituído por um analisador de tempo de vôo
Recebido em 7/11/06; aceito em 10/8/07; publicado na web em 26/2/08
- 1. Vékey, K.; J. Chromatogr., A 2001, 921, 227.
- 2. Ardrey, R. E.; Liquid Chromatography-Mass Spectrometry: An Introdution, Wiley: Huddersfield, 2003.
- 3. Kitson, F. G.; Larsen, B. S.; McEwen, C. N.; Gas Chromatography and Mass Spectrometry - A Pratical Guide, Academic: London, 1996.
- 4. Smeraglia, J.; Baldrey, S. F.; Watson, D.; Chromatographia 2002, 55, 95.
- 5. Granby, K.; Andersen, J. H.; Christensen, H. B.; Anal. Chim. Acta 2004, 520, 165.
- 6. Núñez, O.; Moyano, E.; Galceran, M. T.; Tr AC, Trends Anal. Chem. 2005, 24, 683.
- 7. Bos, S. J.; van Leeuwen, S. M.; Karst, U.; Anal. Bioanal. Chem. 2006, 384, 85.
- 8. Robb, D. B.; Covey, T. R.; Bruins, A. P.; Anal. Chem. 2000, 72, 3653.
- 9. Hager, J. W.; Le Blanc, J. C. Y.; J. Chromatogr., A 2003, 1020, 3.
-
10http://www.chm.bris.ac.uk/ms/theory/cid-fragmentation.html, acessada em Agosto 2006.
» link - 11. Borchers, C.; Parker, C. E.; Deterding, L. J.; Tomer, K. B.; J. Chromatogr., A 1999, 854, 119.
- 12. Aquino Neto, F. R.; Nunes, D. S. S.; Cromatografia: Princípios e Técnicas Afins, Editora Interciência: Rio de Janeiro, 2003.
- 13. Veja, P. V.; Florentino, B. L.; Toxicologia de Alimentos, Centro Nacional de Salud Ambiental: México, 2000.
- 14. Lehotay, S. J.; Hajslová, J.; Tr AC, Trends Anal. Chem. 2002, 21, 686.
- 15. Sorensen, L. K.; Rapid Commun. Mass Spectrom. 2006, 20, 1135.
- 16. Kuklenyik, Z.; Reich, J. A.; Tully, J. S.; Needham, L. L.; Calafat, A. M.; Environ. Sci. Technol. 2004, 38, 3698.
- 17. Taylor, M. J.; Hunter, K.; Hunter, K. B.; Lindsay, D.; Bouhellec, S. L.; J. Chromatogr., A 2002, 982, 225.
- 18. Frenich, A. G.; Vidal, J. L.; López, T. L.; Aguado, S. C.; Salvador, I. M.; J. Chromatogr., A 2004, 1048, 199.
- 19. Arrebola, F. J.; Vidal, J. L. M.; González-Rodríguez, M. J.; Frenich, A. G.; Morito, N. S.; J. Chromatogr., A 2003, 1005, 131.
- 20. Vidal, J. L. M.; Arrebola, F. J.; Mateu-Sánchez, M.; J. Chromatogr., A 2002, 959, 203.
- 21. Arrebola, F. J.; Vidal, J. L. M.; Mateu-Sánchez, M.; Álvarez-Castellón, F. J.; Anal. Chim. Acta 2003, 484, 167.
- 22. Moreno, J. L. F.; Liébanas, F. J. A.; Frenich, A. G.; Vidal, J. L. M.; J. Chromatogr., A 2006, 1111, 97.
- 23. Patel, K.; Fussel, R. J.; Hetmanski, M.; Goodal, D. M.; Keely, B. J.; J. Chromatogr., A 2005, 1068, 289.
- 24. González-Rodríguez, M. J.; Liébanas, F. J. A.; Frenich, A. G.; Vidal, J. L. M.; López, F. J. S.; Anal. Bioanal. Chem. 2005, 382, 164.
- 25. Sannino, A.; Bolzoni, L.; Bandini, M.; J. Chromatogr., A 2004, 1036, 161.
- 26. Ferrer, C.; Gómez, M. J.; García-Reyes, J. F.; Ferrer, I.; Thurman, E. M.; Fernández-Alba, A. R.; J. Chromatogr., A 2005, 1069, 183.
- 27. Fagerquist, C. K.; Lightfield, A. R.; Lehotay, S. J.; Anal. Chem. 2005, 77, 1473.
- 28. Bogialli, S.; Capitolino, V.; Curini, R.; Di Corcia, A.; Nazzari, M.; Sergi, M.; J. Agric. Food. Chem. 2004, 52, 3286.
- 29. Matabudul, D. K.; Conway, B.; Lumley, I.; Sumar, S.; Food Chem. 2001, 75, 345.
- 30. Andersen, W. C.; Roybal, J. E.; Gonzales, S. A.; Turnipseed, S. B.; Pfenning, A. P.; Kuck, L. R.; Anal. Chim. Acta 2005, 529, 145.
- 31. Nicolich, R. S.; Werneck-Barroso, E.; Marques, M. A. S.; Anal. Chim. Acta 2006, 565, 97.
- 32. Mottier, P.; Khong, S. P.; Gremaud, E.; Richoz, J.; Delatour, T.; Goldmann, T.; Guy, P. A.; J. Chromatogr., A 2005, 1067, 85.
- 33. Heller, D. N.; Ngoh, M. A.; Donoghue, D.; Podhorniak, L.; Righter, H.; Thomas, M. H.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2002, 774, 39.
- 34. Mottier, P.; Huré, I.; Gremaud, E.; Guy, P. A.; J. Agric. Food Chem. 2006, 54, 2018.
- 35. Chen, C. Y.; Li, W. J.; Peng, K. Y.; J. Agric. Food Chem. 2005, 53, 8474.
- 36. Draisci, R.; Palleschi, L.; Ferretti, E.; Fuery, A.; James, K. J.; Satake, M.; Yasumoto, T.; J. Chromatogr., A 2000, 871, 13.
- 37. Rundberget, T.; Wilkins, A. L.; J. Chromatogr., A 2002, 964, 189.
- 38. Lau, B. P. Y.; Scott, P. M.; Lewis, D. A.; Kanhere, S. R.; Clérox, C.; Roscoe, V. A.; J. Chromatogr., A 2003, 998, 119.
- 39. Häubl, G.; Berthiller, F.; Krska, R.; Schuhmacher, R.; Anal. Bioanal. Chem. 2006, 384, 692.
- 40. Eppe, G.; Focant, J. F.; Pirard, C.; Pauw, E.D.; Talanta 2004, 63, 1135.
- 41. Hoenicke, K.; Gatermann, R.; Harder, W.; Hartig, L.; Anal. Chim. Acta 2004, 520, 207.
- 42. Bermudo, E.; Moyano, E.; Puignou, L.; Galceran, M.T.; Anal. Chim. Acta 2006, 559, 207.
- 43. Zhang, Y.; Jiao, J.; Ren, Y.; Wu, X.; Zhang, Y.; Anal. Chim. Acta 2005, 551, 150.
- 44. Andrzejewski, D.; Roach, J. A. G.; Gay, M. L.; Musser, S. M.; J. Agric. Food Chem. 2004, 52, 1996.
Datas de Publicação
-
Publicação nesta coleção
16 Jun 2008 -
Data do Fascículo
2008
Histórico
-
Aceito
10 Ago 2007 -
Recebido
07 Nov 2006