Resumo
This work describes a comparative study of the electrocrystallization of Ni and Ni-P on Pt ultramicroelectrodes using chronoamperometric measurements. It was possible to confirm that in all cases a progressive nucleation was the predominant mechanism. Moreover, the application of the Atomistic Theory to the experimental rate of nuclei formation showed that the number of atoms in the critical nucleus was zero, except for Ni-P on Pt at low overpotentials were a value of one was observed. Furthermore, the physical characterisation of the different deposits on Pt by atomic force microscopy allowed observing the coalescence of the hemispherical nuclei of Ni and Ni-P at t max thus confirming the results obtained from the current-time analysis.
electrocrystallization; nickel-phosphorous; Pt ultramicroelectrode
electrocrystallization; nickel-phosphorous; Pt ultramicroelectrode
ARTIGO
Estudo da eletrocristalização de Ni e Ni-P sobre ultramicroeletrodo de platina
Study of electrocrystallization of Ni and Ni-P on platinum ultramicroelectrode
Katlin Ivon Barrios EguiluzI, * * e-mail: kibe@iqsc.usp.br ; Luis Alberto AvacaI; Cláudia Smaniotto BarinII
IInstituto de Química de São Carlos, Universidade de São Paulo, CP 780, 13560-970 São Carlos - SP, Brasil
IICurso de Química Industrial, Universidade Norte do Paraná, Av. Paris, 675, 86041-100 Londrina PR, Brasil
ABSTRACT
This work describes a comparative study of the electrocrystallization of Ni and Ni-P on Pt ultramicroelectrodes using chronoamperometric measurements. It was possible to confirm that in all cases a progressive nucleation was the predominant mechanism. Moreover, the application of the Atomistic Theory to the experimental rate of nuclei formation showed that the number of atoms in the critical nucleus was zero, except for Ni-P on Pt at low overpotentials were a value of one was observed. Furthermore, the physical characterisation of the different deposits on Pt by atomic force microscopy allowed observing the coalescence of the hemispherical nuclei of Ni and Ni-P at tmax thus confirming the results obtained from the current-time analysis.
Keywords: electrocrystallization; nickel-phosphorous; Pt ultramicroelectrode.
INTRODUÇÃO
O estudo da eletrocristalização de níquel em diferentes substratos é de grande interesse devido sua aplicação tecnológica.1 As ligas metálicas amorfas (LMA) de Ni-P e Ni-B possuem características diferenciadas, como dureza, resistência a ataques químicos, aderência, soldabilidade, etc., que as tornam muito adequadas para recobrimentos protetores contra a corrosão, recobrimentos com fins ornamentais, aplicações em dispositivos eletrônicos, entre outros.
Nas últimas décadas, as LMA têm atraído o interesse de muitos pesquisadores, devido as suas propriedades mecânicas,2 magnéticas,3 elétricas4 e, em algumas situações por sua grande resistência contra a corrosão.5 As LMA são normalmente soluções sólidas supersaturadas contendo um metalóide como P e/ou B.
As LMA, ao contrário das ligas cristalinas, caracterizam-se por apresentarem estrutura homogênea, com arranjo desordenado dos átomos que compõem a liga. Esta é a característica fundamental deste material, pois se podem estudar as LMA em uma ampla faixa de composição sem que o material sofra transição de fase.
Em ligas obtidas por deposição autocatalítica ou por eletrodeposição, o metalóide age como um agente redutor do metal, além de servir como agente causador da amorfia, já que a rede de cristalização do metal é diferente da do metalóide e esta situação pode permitir a predominância de uma estrutura amorfa no depósito. Se por um lado o metalóide estabiliza a fase amorfa, em contrapartida, a presença deste altera as propriedades magnéticas, elétricas e mecânicas por doação de elétrons aos orbitais d dos metais.
As propriedades químicas também são alteradas, pois a adição do metalóide pode permitir que a liga adquira um caráter químico mais inerte, uma vez que as LMA não possuem defeitos cristalinos ou interfases nas vizinhanças dos grãos, dificultando a ocorrência de ataques localizados, como acontece com as ligas convencionais cristalinas.
Um aspecto fundamental na eletrodeposição é o estudo dos estágios iniciais na formação de uma nova fase, chamada de eletrocristalização. A formação dos núcleos é, geralmente, promovida pela aplicação de um sobrepotencial. Os mecanismos de nucleação apresentam duas formas limitantes: nucleação instantânea, quando todos os núcleos são formados imediatamente após um apropriado potencial ou, nucleação progressiva, quando os núcleos são formados continuamente seguindo uma cinética de primeira ordem após um salto de potencial.
A eletrocristalização de metais em diferentes substratos continua provocando um grande interesse na eletroquímica moderna, devido à sua importância tecnológica. A cinética de nucleação e o crescimento do primeiro núcleo metálico sobre um substrato são etapas críticas que determinam as propriedades físicoquímicas dos eletrodepósitos e são cruciais no entendimento e controle do processo.6
Dentro destas primeiras etapas (formação dos primeiros núcleos), a determinação do número de átomos necessários para a formação de um aglomerado estável que possa crescer (formação do núcleo crítico) é de grande interesse e em circunstâncias particulares é possível controlar o tamanho, a forma e a energia requerida na formação de agregados estáveis chamados nanopartículas.7
Os mecanismos da eletrocristalização (nucleação) de Ni foram amplamente estudados sobre diversos materiais eletródicos.8-11 Petrovic e colaboradores9 estudaram a nucleação de Ni sobre um arranjo de microeletrodos de carbono. A partir da análise dos transientes de corrente usando as equações do modelo modificado de Sharifker e Mostany, os autores observaram que a eletrocristalização segue uma nucleação progressiva tridimensional e um crescimento controlado pela difusão.
Correia e colaboradores10 estudaram a eletrocristalização de Ni sobre carbono vítreo em solução de cloreto de níquel por cronoamperometria. A análise dos transientes utilizando o modelo de Scharifker e Hills revelou que a nucleação do níquel em ambos os substratos é progressiva e controlada por difusão.
Por outro lado, existem diversos trabalhos que investigaram a eletrodeposição de ligas de Ni-P sobre diversos substratos (carbono vitreo,12 titânio anodizado,13 cobre,14-16 aço inoxidável17 e platina).18 Croussier e colaboradores12 investigaram os mecanismos de deposição da liga amorfa Ni-P sobre carbono vítreo por voltametria cíclica, relatando que o sobrepotencial de nucleação decresce com a velocidade de varredura. A varredura anódica apresentou dois picos bem definidos e associados à dissolução do filme de Ni-P anteriormente depositado. O primeiro pico está associado a depósitos ricos em Ni e o segundo pico a depósitos ricos em P. O primeiro pico é controlado por um processo cinético bastante lento, provavelmente como conseqüência das modificações na composição e pH da solução na interface, produzida pela deposição da liga rica em fósforo.
Djokic17 estudou a eletrodeposição de ligas amorfas como Ni-P, Co-P e Ni-Co-P a partir de soluções ácidas contendo ácido fosforoso como fonte de P. Os resultados obtidos por voltametria linear indicam que a deposição de Ni-P, Co-P e Ni-Co-P ocorre para potenciais mais negativos que 0,70 V vs. eletrodo de calomelano saturado (ECS).
Por outro lado, o estudo dos estágios iniciais (nucleação e crescimento) da eletrodeposição de ligas Ni-P, foi recentemente relatado por Kurowski e colaboradores.19 A deposição da liga foi realizada sobre latão, por cronoamperometria. A análise dos transientes revelou que a nucleação é instantânea e controlada cineticamente. Estudos da morfologia do depósito realizados por microscopia de força atômica (MFA) confirmaram que a nucleação da liga é instantânea com crescimento hemisférico dos centros.
Os ultramicroeletrodos (UME) provaram ser uma ferramenta conveniente para os estudos de eletrodeposição devido às suas pequenas dimensões, permitindo a formação e crescimento de um número reduzido de núcleos, minimizando a interferência de núcleos vizinhos. Um dos maiores problemas na utilização de UME na forma de disco para este tipo de estudo foi a falta de equações que descrevam o processo de nucleação controlada pela difusão esférica. Este efeito foi discutido anteriormente20,21 e impôs a necessidade de desenvolver equações que interpretem corretamente os transientes de corrente em UMEs.
Em um trabalho publicado pelo nosso grupo de pesquisa foi desenvolvido um novo modelo22 para ser usado em estudos de nucleação e crescimento de núcleos hemisféricos controlados pela difusão em UME na forma de disco. A utilização deste modelo envolve a regressão não linear dos dados experimentais com as equações teóricas para a determinação dos parâmetros N (número de núcleos formados por unidade de área em cm2) ou N∞ (taxa de formação de núcleos em cm-2 s-1).
Devido à deficiência de estudos dos estágios iniciais de eletrodeposição da liga Ni-P, o objetivo deste trabalho foi estudar os estágios iniciais dos processos de eletrodeposição da liga Ni-P sobre substrato de Pt e realizar uma comparação com os estágios iniciais de eletrodeposição de Ni. Para isto, foram utilizadas as técnicas de voltametria cíclica, cronoamperometria e, finalmente, a morfologia dos depósitos nas suas fases iniciais foi analisada por MFA.
PARTE EXPERIMENTAL
A solução eletrolítica para a eletrodeposição de Ni consistia em um banho contendo NiSO4. 6H2O 0,1 mol L-1, CH3COONa 0,15 mol L-1, e para a liga Ni-P foram usadas soluções semelhantes às usadas na deposição autocatalítica de Ni-P para verificar o possível efeito deste processo nas características de nucleação e crescimento da nova fase metal-metalóide. Assim, o banho consistia em NiSO4. 6H2O 0,1 mol L-1, CH3COONa 0,15 mol L-1, NaH2PO2 0,1 mol L-1 ajustando-se para ambos o pH até 4,8 com CH3COOH. As soluções eletrolíticas foram preparadas a partir de reagentes Merck P.A., com água purificada por um sistema Milli-Q (Millipore, Inc.). Antes da realização de medidas eletroquímicas, borbulhava-se N2 superseco (White Martins) durante 15 min. O N2 era continuamente injetado no compartimento da cela, acima do nível do eletrólito durante a realização dos experimentos, para evitar a entrada de oxigênio na cela.
Para a realização das medidas eletroquímicas foi utilizada uma cela de um compartimento com dois eletrodos, o ultramicroeletrodo consistia em um fio de platina (99,99% Degussa) com Φ = 50 µm, fabricado em nosso laboratório por isolamento do respectivo fio em vidro, e utilizou-se como sistema auxiliar/referência o ECS, todos os potenciais mostrados neste estudo estão referidos a este eletrodo de referência. Para as medidas eletroquímicas foi utilizado um potenciostato/galvanostato modelo 273(EG & G PAR), acoplado a um microcomputador tipo IBM-PC modelo 486 com software M270 (EG & G PAR).
As imagens de MFA foram obtidas em um microscópio de tunelamento e força atômica TM AFM Explorer da TopoMetrix®. As medidas de energia dispersiva de raios X (EDX) foram realizadas em um microscópio eletrônico de varredura LEO modelo 440 (Laica-Zeiss) acoplado a um microanalisador Oxford modelo 7060.
RESULTADOS E DISCUSSÃO
A resposta voltamétrica para a eletrodeposição de Ni e Ni-P em UME de platina a 100 mV s-1 pode ser visualizada na Figura 1. Na varredura catódica observa-se que a deposição de Ni na ausência e na presença do agente amorfizante apresenta o cruzamento dos ramos anódico e catódico, que está associado a um aumento da área eletroativa na superfície do eletrodo, sendo indicativo de um processo de nucleação e crescimento.23
A deposição do Ni inicia-se em aproximadamente -500 mV, apresentando um pico catódico de deposição em torno de -750 mV. Os valores de carga associados à dissolução de Ni na ausência de fósforo são consideravelmente pequenos se comparados à carga de deposição. Isto ocorre devido ao fato do Ni sofrer um processo de passivação, conforme descrito na literatura.24
O comportamento voltamétrico do Ni na presença do agente amorfizante é visualmente alterado, sendo que a dissolução do depósito obtido na presença do agente amorfizante apresenta um máximo de corrente em aproximadamente 1000 mV, característico da espécie Ni-P.12
Por outro lado, o inicio da reação de desprendimento de hidrogênio, estudada sobre o UME de Pt, ocorre em potencias mais negativos que -1000 mV (curva não apresentada). Assim, a deposição da liga Ni-P acontece sem interferências, fato este que é corroborado pela relação das cargas anódica e catódica, que apresenta um valor de ~1.
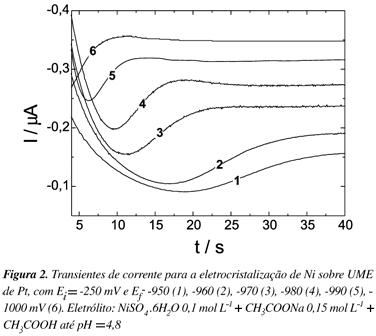
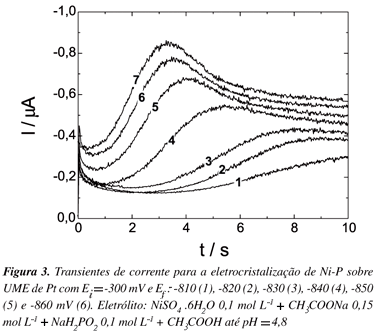
Utilizando os dados da voltametria cíclica, realizou-se o estudo da eletrodeposição potenciostática de Ni e Ni-P. Nas Figuras 2 e 3 podem-se visualizar a família de transientes obtida para a nucleação de Ni e Ni-P sobre eletrodo de Pt, respectivamente. O potencial inicial dos saltos foi -250 mV para Ni e -300 mV para Ni-P, e o potencial final estava entre -950 e -1000 mV para Ni e entre -810 até -870 mV para Ni-P.
Os transientes de corrente obtidos apresentam um único máximo de corrente, comportamento comumente encontrado para processos de nucleação e crescimento.
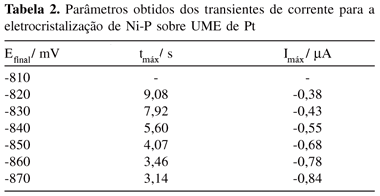
Os parâmetros característicos obtidos dos transientes das Figuras 2 e 3 (tempo máximo e corrente máxima) são apresentados nas Tabelas 1 e 2, onde se observa que, para valores de potenciais mais negativos ocorre um decréscimo nos valores de tempo e um acréscimo nos valores de corrente, indicando que o processo de nucleação é mais rápido em potenciais mais negativos.
Os transientes de corrente foram analisados de acordo com as equações do modelo teórico para a nucleação sobre UME em forma de disco, desenvolvido em nossos laboratórios22 para processos instantâneos (Equação 1) e progressivos (Equação 2) controlados por difusão.

onde, n é o número de elétrons, F é a constante de Faraday, D é o coeficiente de difusão, c∞ é a concentração das espécies em solução, r é o raio do eletrodo, t é o tempo, N o número de núcleos formados por unidade de área (cm-2), AN∞ é a taxa de formação de núcleos em cm-2 s-1 e k e k' são parâmetros que dependem das condições experimentais e são dados por:

onde, M é o peso molecular do íon metálico e ρ é a densidade do metal.
A reprodução matemática dos transientes experimentais foi realizada ajustando as Equações 1 e 2 aos dados experimentais usando o procedimento de regressão não linear do programa Microcal Origin®. As variáveis usadas para realizar a regressão foram D, N e AN∞ para cada tipo de nucleação.
Nas Figuras 4 e 5 apresenta-se o comportamento dos transientes experimentais com relação às curvas teóricas de Ni e Ni-P, respectivamente, tanto para nucleação progressiva como para a instantânea. Para fins de simplificação, encontram-se representadas apenas as curvas correspondentes aos potenciais de salto equivalentes a -950 e -980 mV para Ni e -810 e -850 mV para Ni-P, sendo estes representativos do conjunto. Pode se observar na mesma figura uma grande concordância dos dados experimentais com a curva teórica progressiva, sendo semelhante para todos os potenciais de salto.
Pelos ajustes realizados utilizando a Equação 2 obtiveram-se os valores para a taxa de formação de núcleos, AN∞, que podem ser visualizados na Tabela 3 para deposição de Ni e na Tabela 4 para a deposição de Ni-P, bem como o valor do coeficiente de difusão D. Os valores de AN∞ obtidos, em relação aos potenciais de salto, apresentam o comportamento esperado, com o valor de AN∞ crescendo com o aumento do potencial aplicado. O valor do coeficiente de difusão obtido pelos ajustes foi 2,8 × 10-6 cm2s-1 para Ni e de 2,9 × 10-6 cm2s-1 para a liga Ni-P.
Teoria atomística
Com a finalidade de observar a relação entre AN∞ e o potencial de salto é necessário considerar a teoria atomística da eletrocristalização, que analisa a formação do núcleo crítico. De acordo com esta teoria, a derivada parcial da taxa de formação de núcleos com respeito ao potencial final dos saltos potenciostáticos é expressa pela Equação 5:25

onde n* é o número de átomos no núcleo crítico, α é o coeficiente de transferência eletroquímico e as demais constantes mantêm os significados usuais.
Considerando a Equação 5 e o coeficiente angular da reta obtida nas Figuras 6 e 7, (64,53 mV-1 para Ni e 90,1 mV-1 para Ni-P) é possível calcular o valor de n* para a nucleação de Ni e da liga Ni-P. Isto foi realizado assumindo que o valor de α seja igual a 0,5, resultando em n* = 0,3 para Ni e 0,2 para a liga Ni-P. Os valores dos coeficientes de correlação das retas foram de 0,99833 para Ni e de 0,97519 para a liga Ni-P.
Os resultados assim obtidos sugerem, sob estas condições, a presença de zero átomos de Ni e da liga Ni-P no núcleo crítico. Este valor já foi obtido para a nucleação de Ni sobre carbono,9 chumbo sobre carbono vítreo,26 cobre em paládio27 e platina sobre tungstênio.28 Em termos da teoria atomística dentro do intervalo de potencial investigado, zero átomos no núcleo crítico quer dizer que um simples átomo adsorvido em um sítio ativo do substrato forma um cluster estável de um átomo que pode crescer irreversivelmente nesse potencial.29
Como a reta teve um valor de coeficiente de correlação baixo para a liga Ni-P, realizou-se o cálculo de n* tomando primeiro os valores de potencial de salto menores e, posteriormente, os valores maiores, como se pode observar nas Figuras 8 e 9. O valor de n* calculado pelo coeficiente angular da reta (145,35 mV-1) da Figura 8 para valores de potenciais menores (-810, -820, -830 mV) foi 0,8, sugerindo a presença de 1 átomo no núcleo crítico. Já para valores de potenciais maiores (-850, -860, -870 mV) o valor do coeficiente angular da reta da Figura 9 foi 42,46 V-1 obtendo-se um valor de zero para n* e, portanto, zero átomos no núcleo crítico.
Estudos morfológicos
Para a observação dos aspectos morfológicos dos eletrodepósitos de Ni e Ni-P, foram realizados analises ex-situ por MFA e a composição superficial foi analisada por EDX. Para a caracterização morfológica utilizaram-se depósitos obtidos usando a técnica de cronoamperometria com tempo de deposição equivalente ao tempo de máximo dos transientes de corrente (condição necessária para observar a coalescência dos núcleos).
Na Figura 10A tem-se a imagem de MFA, em três dimensões, para o substrato platina, utilizado como "branco" para comparação com as imagens obtidas para as superfícies contendo os eletrodepósitos de Ni e Ni-P. A Figura 10B apresenta a imagem de MFA para Ni sobre platina, com um potencial de salto de -990 mV e um tempo de deposição de 12 s, que corresponde ao máximo de corrente do transiente. Pode-se observar a existência de núcleos hemisféricos com diâmetros variáveis, confirmando um processo de nucleação progressiva controlado por difusão como visto na Figura 4.

Da mesma forma, na Figura 10C pode-se visualizar os núcleos de Ni-P crescidos com potencial inicial igual a -200 mV, potencial de salto de -840 mV, e um tempo de deposição de 5 s. Os núcleos da liga Ni-P apresentam forma hemisférica com diâmetros variáveis distribuídos sobre o substrato de platina, corroborando os resultados obtidos por meio dos ajustes das equações para o processo de nucleação progressiva, mostrado na Figura 5. Adicionalmente, a análise por EDX para estes depósitos confirmou a presença de Ni e Ni-P nos depósitos. Assim, as porcentagens atômicas obtidas para a liga Ni-P foram de 57,40% para o Ni e de 42,60% para o P.
CONCLUSÕES
Os resultados obtidos para a análise dos transientes de Ni e NiP em UMEs revelam que a nucleação ocorre em forma progressiva em ambos os casos. A dependência da taxa de variação dos núcleos com o potencial final dos saltos potenciostáticos para Ni e Ni-P foi analisada pela Teoria Atomística. Os resultados sugerem a presença de zero átomos no núcleo crítico para os dois casos, com exceção do Ni-P sobre platina em baixos potenciais de salto onde foi observado 1 átomo no núcleo crítico. Entretanto, não foi evidenciada a ocorrência do processo de deposição autocatalítica.
A caracterização física dos filmes de Ni e NiP realizada por MFA confirmou que os processos de nucleação de Ni e Ni-P acontecem de forma progressiva. Por outro lado, a análise por EDX confirmou a presença de Ni e Ni-P nos depósitos.
AGRADECIMENTOS
Ao apoio financeiro do CNPq, ao Dr. G. R. Salazar-Banda do Instituto de Química de São Carlos - Universidade de São Paulo pela sua valiosa contribuição a este trabalho.
REFERÊNCIAS
1. Clark, D.; Wood, D.; Erb, U.; NanoStructured Materials 1997, 9, 755.
2. Wu, F-B.; Duh, J-G.; Thin Solid Films 2003, 441, 165.
3. Tachev, D.; Georgieva, J.; Armyanov, S.; Electrochim. Acta 2001, 47, 359.
4. Cheng, P. L.; Leung, S. Y. Y.; Law, T. W.; Liu, C. K.; Chong, I. T.; Lam, D. C. C.; Proceedings of the 53 rd Electronic Components and Technology Conference 2003, 156.
5. Bai, A.; Chuang, P-Y.; Hu, C-C.; Mater. Chem. Phys. 2003, 82, 93.
6. Jafarian, M.; Mahjani, M. G.; Gobal, F.; Danaee, E.; J. Electroanal. Chem. 2006, 588, 190.
7. Palomar-Pardavé, M.; Gonzéles, I.; Batina, N.; Resumos do XIII Congreso da Sociedade Iberoamericana de Eletroquimica, Viña del Mar, Chile, 1998.
8. Grujicic, D.; Pesic, B.; Electrochim. Acta 2006, 51, 2678.
9. Petrovic, Z.; Metikos-Hukovic, M.; Grubac, Z.; Omanovic, S.; Thin Solid Films 2006, 513, 193.
10. Correia, A. N.; Machado, S. A. S.; Avaca, L. A.; J. Electroanal. Chem. 2000, 488, 110.
11. Gómez, E.; Müller, C.; Pollina, R.; Sarret, M.; Vallés, E.; J. Electroanal. Chem. 1992, 333, 47.
12. Crousier, J.; Hanane, Z.; Crousier, J-P.; Electrochim. Acta 1993, 38, 261.
13. Delplancke, J. L.; Winand, R.; Dierickx, M.; Lifschitz, L.; J. Electrochem. Soc. 1993, 140, 2800.
14. Morikawa, T.; Nakade, T.; Yukoi, M.; Fukumoto, Y.; Iwakura, C.; Electrochim. Acta 1997, 42, 115.
15. Bonino, J. P.; Bruet-Hotellaz, S.; Pouderoux, P.; Rousset, A.; J. Appl. Electrochem. 1997, 27, 1193.
16. Mahalingam, T.; Raja, M.; Thanikaikarasan, S.; Sanjeeviraja, C.; Velumani, S.; Hosun, M.; Yong, D. K.; Mater. Charact. 2007, 58, 800 .
17. Djokic, S. S.; J. Electrochem. Soc. 1999, 146, 1824.
18. Ordine, A, P.; Díaz, S, L.; Margarit, I. C. P.; Barcia, O. E.; Mattos, O. P.; Electrochim. Acta 2006, 51, 1480.
19. Kurowski, A.; Schultze, J. W.; Staikov, G.; Electrochem. Commun. 2002, 4, 565.
20. Wightman, R. M.; Wipf, D. O. Em Electroanalytical Chemistry; Bard, A. J., ed.; Marcel Dekker: New York, 1989, p. 267.
21. Aoki, K.; Osteryoung, J.; J. Electroanal. Chem. 1984, 160, 335.
22. Correia, A. N.; Machado, S. A. S.; Sampaio, J. C. V.; Avaca, L. A.; J. Electroanal. Chem. 1996, 407, 37.
23. Correia, A. N.; Machado, S. A. S.; Electrochim. Acta 2000, 45, 1733.
24. Lain, M. J.; Pletcher, D.; Electrochim. Acta 1987, 32, 99.
25. Milchev, A.; Tsakova, V.; J. Appl. Electrochem. 1990, 20, 301.
26. Mostany, J.; Mozota, J.; Scharifker, B. R.; J. Electroanal. Chem. 1984, 177, 25.
27. Rigano, P. M.; Mayer, C.; Chierchie, T.; Electroanal. Chem. 1988, 248, 219.
28. Kelaidopoulou, A.; Kokkinidis, G.; Milchev, A.; J. Electroanal. Chem. 1998, 444, 195
29. Yang, M.; Hu, Z.; J. Electroanal. Chem. 2005, 583, 46.
Recebido em 6/8/07; aceito em 26/10/07; publicado na web em 27/6/08
- 1. Clark, D.; Wood, D.; Erb, U.; NanoStructured Materials 1997, 9, 755.
- 2. Wu, F-B.; Duh, J-G.; Thin Solid Films 2003, 441, 165.
- 3. Tachev, D.; Georgieva, J.; Armyanov, S.; Electrochim. Acta 2001, 47, 359.
- 4. Cheng, P. L.; Leung, S. Y. Y.; Law, T. W.; Liu, C. K.; Chong, I. T.; Lam, D. C. C.; Proceedings of the 53 rd Electronic Components and Technology Conference 2003, 156.
- 5. Bai, A.; Chuang, P-Y.; Hu, C-C.; Mater. Chem. Phys. 2003, 82, 93.
- 6. Jafarian, M.; Mahjani, M. G.; Gobal, F.; Danaee, E.; J. Electroanal. Chem 2006, 588, 190.
- 7. Palomar-Pardavé, M.; Gonzéles, I.; Batina, N.; Resumos do XIII Congreso da Sociedade Iberoamericana de Eletroquimica, Viña del Mar, Chile, 1998.
- 8. Grujicic, D.; Pesic, B.; Electrochim. Acta 2006, 51, 2678.
- 9. Petrovic, Z.; Metikos-Hukovic, M.; Grubac, Z.; Omanovic, S.; Thin Solid Films 2006, 513, 193.
- 10. Correia, A. N.; Machado, S. A. S.; Avaca, L. A.; J. Electroanal. Chem 2000, 488, 110.
- 11. Gómez, E.; Müller, C.; Pollina, R.; Sarret, M.; Vallés, E.; J. Electroanal. Chem 1992, 333, 47.
- 12. Crousier, J.; Hanane, Z.; Crousier, J-P.; Electrochim. Acta 1993, 38, 261.
- 13. Delplancke, J. L.; Winand, R.; Dierickx, M.; Lifschitz, L.; J. Electrochem. Soc 1993, 140, 2800.
- 14. Morikawa, T.; Nakade, T.; Yukoi, M.; Fukumoto, Y.; Iwakura, C.; Electrochim. Acta 1997, 42, 115.
- 15. Bonino, J. P.; Bruet-Hotellaz, S.; Pouderoux, P.; Rousset, A.; J. Appl. Electrochem 1997, 27, 1193.
- 16. Mahalingam, T.; Raja, M.; Thanikaikarasan, S.; Sanjeeviraja, C.; Velumani, S.; Hosun, M.; Yong, D. K.; Mater. Charact. 2007, 58, 800 .
- 17. Djokic, S. S.; J. Electrochem. Soc. 1999, 146, 1824.
- 18. Ordine, A, P.; Díaz, S, L.; Margarit, I. C. P.; Barcia, O. E.; Mattos, O. P.; Electrochim. Acta 2006, 51, 1480.
- 19. Kurowski, A.; Schultze, J. W.; Staikov, G.; Electrochem. Commun. 2002, 4, 565.
- 20. Wightman, R. M.; Wipf, D. O. Em Electroanalytical Chemistry; Bard, A. J., ed.; Marcel Dekker: New York, 1989, p. 267.
- 21. Aoki, K.; Osteryoung, J.; J. Electroanal. Chem. 1984, 160, 335.
- 22. Correia, A. N.; Machado, S. A. S.; Sampaio, J. C. V.; Avaca, L. A.; J. Electroanal. Chem. 1996, 407, 37.
- 23. Correia, A. N.; Machado, S. A. S.; Electrochim. Acta 2000, 45, 1733.
- 24. Lain, M. J.; Pletcher, D.; Electrochim. Acta 1987, 32, 99.
- 25. Milchev, A.; Tsakova, V.; J. Appl. Electrochem. 1990, 20, 301.
- 26. Mostany, J.; Mozota, J.; Scharifker, B. R.; J. Electroanal. Chem 1984, 177, 25.
- 27. Rigano, P. M.; Mayer, C.; Chierchie, T.; Electroanal. Chem 1988, 248, 219.
- 28. Kelaidopoulou, A.; Kokkinidis, G.; Milchev, A.; J. Electroanal. Chem 1998, 444, 195
- 29. Yang, M.; Hu, Z.; J. Electroanal. Chem 2005, 583, 46.
Datas de Publicação
-
Publicação nesta coleção
12 Set 2008 -
Data do Fascículo
2008
Histórico
-
Aceito
26 Out 2007 -
Recebido
06 Ago 2007