Resumo
Structure and first hyperpolarizability for a series of armchair a(5,5) chemically modified carbon nanotubes (CNT) were calculated at semiempirical and density functional levels of theory. The 4,4´-substituted stilbenes were selected as chromophore with substituents at position 4´ set to X=NO2, H, Cl, OH and NH2. The calculated values for static first hyperpolarizability (β) were almost linearly dependent on the electronic effect of the group X, increasing from NO2 to NH2. At DFT level the effect of inserting the chromophore in the CNT surface was to enhance the β value up to 70% relative to the free 4,4´-substituted stilbene.
SWNT; NLO; ab-initio
SWNT; NLO; ab-initio
ARTIGO
Estudo teórico de propriedades ópticas não-lineares de nanotubos de carbono de parede única quimicamente modificados
Theoretical analysis of non-linear optical properties for chemically modified single wall carbon nanotubes
Antônio M. Da Silva Jr.I; Geórgia M. A. JunqueiraI; Hélio F. Dos SantosI,** e-mail: helio.santos@ufjf.edu.br; Ana Cláudia M. CarvalhoII
IDepartamento de Química, Instituto de Ciências Exatas, Universidade Federal de Juiz de Fora, 36036-330 Juiz de Fora - MG, Brasil
IIDepartamento de Física e Química, Instituto de Ciências Exatas, Universidade Federal de Itajubá, 37500-000 Itajubá - MG, Brasil
ABSTRACT
Structure and first hyperpolarizability for a series of armchair a(5,5) chemically modified carbon nanotubes (CNT) were calculated at semiempirical and density functional levels of theory. The 4,4´-substituted stilbenes were selected as chromophore with substituents at position 4´ set to X=NO2, H, Cl, OH and NH2. The calculated values for static first hyperpolarizability (β) were almost linearly dependent on the electronic effect of the group X, increasing from NO2 to NH2. At DFT level the effect of inserting the chromophore in the CNT surface was to enhance the β value up to 70% relative to the free 4,4´-substituted stilbene.
Keywords: SWNT; NLO; ab-initio.
INTRODUÇÃO
Descobertos em 1991 por Sumio Iijima,1 os nanotubos de carbono (CNTs) vêm sendo objeto de um extensivo número de trabalhos, sob diversas abordagens, tanto teóricas2-4 quanto experimentais.5-7 Trata-se de uma forma alotrópica do carbono, na qual arranjos hexagonais destes átomos, com hibridização nominal sp2, se dispõem ao longo de superfícies cilíndricas de diâmetros da ordem de nanômetros e comprimentos da ordem de micrômetros.8 Podem ser observadas estruturas de múltiplas camadas concêntricas (MWNTs) ou estruturas de uma única camada (SWNTs), sendo que as extremidades podem ser constituídas por formas semi-esféricas (hemi-fulerenos), formas cônicas ou poliédricas.9 Para melhor entendimento das estruturas, utiliza-se um modelo no qual se admite que cada camada dos tubos corresponda a uma folha de grafeno envolta a uma direção axial.10 Considerando a rede bidimensional do grafeno, pode-se descrevê-la através dos vetores quiral  h e de translação
h e de translação  . O vetor quiral conecta dois pontos cristalograficamente equivalentes ao longo da circunferência do tubo, sendo este definido em termos dos parâmetros de rede do grafeno
. O vetor quiral conecta dois pontos cristalograficamente equivalentes ao longo da circunferência do tubo, sendo este definido em termos dos parâmetros de rede do grafeno  1 e 2 por meio da relação h = n
1 e 2 por meio da relação h = n
 m) com n > m > 0. O vetor translação, ortogonal ao vetor quiral, indica a direção axial dos tubos, na qual as estruturas podem ser formadas através da projeção de suas respectivas celas unitárias.10
m) com n > m > 0. O vetor translação, ortogonal ao vetor quiral, indica a direção axial dos tubos, na qual as estruturas podem ser formadas através da projeção de suas respectivas celas unitárias.10
A busca de relações entre estrutura e propriedades tem sido significativamente avaliada para nanotubos de carbono, visando possíveis aplicações destes em diversas áreas da ciência.11 Um exemplo químico de relação estrutura-propriedade pode ser verificado na reatividade de tais espécies, que é dependente de dois fatores:12 ângulo de piramidalização (θp = (θσπ - 90)º, onde θσπ é o ângulo compreendido entre um orbital σ e um orbital π em um mesmo átomo de carbono) e ângulo de "falta de alinhamento" (missaligment) (ângulo entre dois orbitais π presentes em átomos de carbono adjacentes). Ambos os fatores variam inversamente com o diâmetro dos tubos e é esperado que tubos de menor diâmetro sejam mais reativos que tubos de maior diâmetro. Sendo as propriedades dependentes da estrutura, pode-se supor que a modificação racional desta possa levar à modulação de uma determinada propriedade. A manipulação de nanotubos de carbono vem sendo amplamente avaliada,13 sendo relatados vários tipos de interações, tais como processos físicos e químicos. Tais interações se fazem importantes no sentido de possibilitar, por exemplo, a dispersão das espécies.14
Nos últimos anos a busca por materiais com propriedades ópticas não-lineares (NLO)15 tem crescido significativamente devido ao interesse em potenciais aplicações destes em computação óptica, telecomunicações e processamento de sinais. Considerando a ampla possibilidade de funcionalização química dos nanotubos de carbono (tais como micro e macro moléculas13), pode-se intuir que estes possam atuar como suporte para grupos que possuam respostas ópticas não-lineares e assim serem aplicados na construção de materiais para dispositivos ópticos. Neste contexto, os derivados do estilbeno (STB, Figura 1a) mostram-se relevantes para tal propriedade, podendo conter diferentes substituintes X, Y.16 Geralmente, apresentam propriedades NLO espécies com elevada assimetria eletrônica, e para tal os pares de substituintes X,Y geralmente são formados por uma espécie doadora e outra aceptora de elétrons,15 dando origem aos chamados compostos push-pull.

A literatura ainda apresenta um número modesto de trabalhos, tanto teóricos quanto experimentais, para propriedades NLO de nanotubos de carbono.17-22 Guo e colaboradores21 relataram recentemente um estudo ab initio sistemático de estrutura e propriedades elétricas de uma série de nanotubos de carbono, incluindo diferentes quiralidades e diâmetros. Os resultados obtidos,21 dentro do formalismo da teoria do funcional de densidade (DFT), sugerem que tubos quirais de pequeno diâmetro (~5 Å) apresentam potencial para aplicação em NLO, como, por exemplo, geração do segundo harmônico (SHG). Outro estudo teórico relevante foi relatado por Sun e colaboradores,22 no qual investigaram o efeito do tamanho do tubo e tipo de extremidade (aberta ou fechada) nos espectros de geração de harmônicos de altas ordens (HHG) para tubos armchair (5,5) e zigzag (9,0). Os resultados mostraram que o fechamento do tubo com hemi-fulerenos afeta de forma expressiva as propriedades ópticas, ao passo que tubos de cadeia finita apresentam características ópticas semelhantes aos tubos de cadeia infinita, sendo essa conclusão mais evidente para tubos armchair. Os trabalhos citados anteriormente descrevem estudos de nanotubos isolados (não modificados), entretanto, pouco tem sido feito com relação ao cálculo de propriedades moleculares considerando nanotubos de carbono quimicamente modificados. Portanto, no presente trabalho alguns derivados do STB foram considerados como ligantes (Figura 1b) para funcionalização covalente de um SWNT armchair (5,5) (a(5,5)) e avaliação de suas propriedades NLO através da utilização de metodologias teóricas. Cálculos semi-empíricos e ab initio (DFT) foram realizados para a obtenção dos elementos do tensor primeira hiperpolarizabilidade βijk. Os objetivos são verificar a adequação de uma metodologia seqüencial para obtenção de geometrias de equilíbrio e respostas NLO para um SWNT a(5,5) isolado e funcionalizado, bem como obter evidências quantitativas de relação entre a estrutura e as propriedades ópticas de interesse. Os resultados obtidos são fundamentais para a definição da metodologia, visando estudos mais amplos incluindo sistemas maiores.
METODOLOGIA TEÓRICA
Para a descrição de SWNTs, diversos autores vêm obtendo geometrias de equilíbrio através de métodos semi-empíricos utilizados isoladamente23 ou em cálculos híbridos (p.e. ONIOM).24 Encontram-se também, para estruturas de menor porte, cálculos ab initio efetuados no nível de teoria Hartree-Fock (HF)25 e teoria do funcional de densidade (DFT),26 não sendo citada uma comparação sistemática entre tais métodos. No presente estudo, uma avaliação da metodologia foi feita para a descrição da estrutura do nanotubo de carbono considerado (a(5,5)), utilizando a aproximação semi-empírica PM3,27 o nível de teoria HF,28 com as funções de base CEP-4G,29 STO-3G30 e 6-31G(d)31 e a DFT32 com os funcionais PBE33 e B3LYP,34 ambos com a função de base 6-31G(d). As geometrias dos derivados do STB (Figura 1b) e dos nanotubos modificados foram otimizadas somente no nível semi-empírico PM3 e utilizadas para o cálculo das propriedades ópticas de interesse.
As propriedades ópticas não-lineares foram obtidas através de cálculos de simulação da geração do segundo harmônico (SHG), utilizando o método coupled perturbed Hartree-Fock (CPHF)35 no nível de teoria DFT-SVWN/6-31G(d)//PM3, implementado no pacote computacional Gaussian-03,36 e time-dependent Hartree-Fock (TDHF),37 utilizando a parametrização semi-empírica PM3, como implementado no pacote computacional MOPAC-07.38 Para o método CPHF foi considerada a aproximação estática (freqüência da radiação incidente nula), já para o método TDHF foi considerada a aproximação dinâmica (cálculo da propriedade incluindo sua dependência em relação à freqüência do campo incidente). No nível DFT, os cálculos foram feitos para a energia do campo ħω = 0,00eV e no nível PM3 os cálculos foram também realizados considerando ħω = 0,65eV (1.910 nm).
A aplicabilidade de métodos teóricos para o cálculo de propriedades NLO tem sido avaliada por alguns autores.39-42 Champagne e colaboradores39 realizaram um trabalho sistemático utilizando diferentes funcionais da DFT para a previsão de polarizabilidade (α) e hiperpolarizabilidades (β e γ) de sistemas conjugados. Os resultados mostraram, de forma geral, um comportamento não sistemático entre os diferentes funcionais com o aumento da cadeia polimérica, fornecendo valores de β superestimados em relação aos cálculos pós-HF (p.e. MP2). Outra conclusão importante desse estudo é a pequena influência da base de funções nos resultados, sendo o esquema padrão 6-31G adequado para a maioria das moléculas estudadas. Mais recentemente, Yang et al.40 compararam valores teóricos e experimentais de β e γ para moléculas aromáticas pequenas, incluindo p-nitro-anilina e uma esquaraina. Os valores calculados apresentaram uma boa concordância com os dados experimentais, independente do funcional utilizado, sugerindo o uso de funcionais locais (LDA) como alternativas para o cálculo de propriedades NLO de sistemas maiores. Resultados semelhantes foram descritos por Davidson41 e Isborn,42 mostrando que a metodologia empregada no presente estudo é adequada para a dimensão das moléculas estudadas.
Diz-se que uma determinada molécula apresenta uma propriedade óptica não-linear quando na expansão das componentes cartesianas do seu momento de dipolo elétrico induzido (µi, com i = x, y e z), em função do campo elétrico ( ), ao redor de um campo elétrico nulo, os coeficientes simbolizados por letras gregas (β, γ, ...) tiverem magnitudes suficientemente elevadas (Equação 1).15
), ao redor de um campo elétrico nulo, os coeficientes simbolizados por letras gregas (β, γ, ...) tiverem magnitudes suficientemente elevadas (Equação 1).15

Os termos αij, βijk e γijkl são denominados polarizabilidade, primeira e segunda hiperpolarizabilidade, respectivamente. Estas são grandezas tensoriais, sendo a primeira hiperpolarizabilidade (β) um tensor com 27 componentes βijk (i, j, k = x, y, z).43 Geralmente, os elementos diferentes de zero são reduzidos em função de aspectos de simetria.44 Nos cálculos PM3, como implementado no programa MOPAC-07, βijk = βikj, fornecem, portanto, 18 componentes distintas do tensor. No nível DFT, apenas 10 elementos são calculados, sendo βij = βji. Os tensores cartesianos (βi) podem ser definidos em função de seus elementos tensoriais como na Equação 2.45

Com os tensores cartesianos pode-se calcular a primeira hiperpolarizabilidade média (<β>) através da Equação 3.

Esta é a propriedade avaliada neste trabalho para a série de moléculas consideradas.
RESULTADOS E DISCUSSÃO
Propriedades estruturais
A partir da escolha de um SWNT a(5,5), com suas extremidades saturadas com átomos de hidrogênio, buscou-se inicialmente avaliar qual seria uma extensão razoável para a estrutura que representasse o sistema real, tendo em vista que suas extensões reais são da ordem de micrômetros (1 µm = 10-6 m), e para cálculos quânticos seriam desejáveis estruturas da ordem de angstrons (1 Å = 10-10 m). Para tal, tomou-se como objeto de estudo a série a(5,5) C30+10kH20, com k inteiro variando de 0 a 9, a exemplo da metodologia utilizada por Bettinger46 para avaliar a influência do comprimento de uma estrutura a(5,5) em suas energias de reação de adição de flúor e metileno através de cálculos DFT. Com isto, a extensão das estruturas foi variada de 3 a 13 Å. Como discutido por Saito e Dresselhaus,47 as celas unitárias de nanotubos aquirais, armchair (n,n) e zigzag (n,0), pertencem aos grupos pontuais Dnh para n par e Dnd para n ímpar. Portanto, a cela unitária da estrutura a(5,5) pertence ao grupo pontual D5d. Porém, a sua repetição periódica para gerar as estruturas faz com que a espécie pertença aos grupos espaciais D5h para k zero ou par, devido à presença de um plano especular horizontal (perpendicular à direção axial do tubo), e D5d para k ímpar, devido à ausência deste plano. Uma referência recente traz uma abordagem mais ampla e geral para a classificação de CNTs quanto à simetria,48 considerando sistemas de dimensões infinitas. Essa classificação é importante na previsão de propriedades dependentes da simetria, entretanto, nos cálculos de propriedades NLO essa informação não é introduzida explicitamente, sendo o valor da grandeza dependente da estrutura molecular tridimensional. É também oportuno ressaltar que os formalismos gerais para a classificação de simetrias dos CNTs são fundamentados em razões comprimento/diâmetro > 104, o que, evidentemente, está além das dimensões das moléculas estudadas no presente trabalho.
Utilizando os diferentes níveis de teoria citados na seção de Metodologia teórica, PM3, HF/(CEP-4G, STO-3G e 6-31G(d)) e (PBE e B3LYP)/6-31G(d), otimizaram-se as estruturas para k variando de 0 a 9 e observaram-se as distâncias médias carbono-carbono (rC-C), como demonstrado na Figura 2.
Pode-se observar que, com exceção do método HF/CEP-4G, todas as aproximações teóricas mostram valores próximos para as distâncias médias de ligação, variando, em média, de no máximo 0,02 Å para cada valor de k. Os valores fornecidos pelo método HF/CEP-4G mostraram-se superestimados em relação aos demais, sendo superiores a 1,50 Å. Em SWNTs, as ligações possuem um caráter intermediário entre uma ligação simples e uma dupla, sendo em média de 1,44 Å.47 Os valores fornecidos pelo método HF/CEP-4G encontram-se mais próximos das distâncias C-C no diamante, de 1,54 Å,47 no qual existem apenas ligações simples, o que é contextualmente inconsistente.
A ampliação da escala para o eixo das ordenadas na Figura 2 permite observar um comportamento sistemático, no qual as distâncias médias de ligação C-C tendem para um valor limite quando o número de carbonos e, conseqüentemente, a extensão da estrutura são aumentados (Figura 1S, material suplementar).
Analisando as distâncias C-C com o inverso do número de carbono (1/nC), pôde-se fazer uma estimativa dos valores de rC-C quando nC tende ao infinito (rC-C (nC
 )). Estas extrapolações para os diferentes métodos estão contidas na Tabela 1, na qual se pode observar que o método PM3 é significativamente equiparável aos funcionais PBE e B3LYP com a função de base estendida 6-31G(d). O valor absoluto para a distância rC-C (nC
)). Estas extrapolações para os diferentes métodos estão contidas na Tabela 1, na qual se pode observar que o método PM3 é significativamente equiparável aos funcionais PBE e B3LYP com a função de base estendida 6-31G(d). O valor absoluto para a distância rC-C (nC  ) obtido com o método semi-empírico destoa em apenas 0,01 Å em relação às distancias fornecidas pelos funcionais e mostrou parâmetros de ajuste linear, tais como o quadrado do coeficiente de correlação (R2=0,920) e desvio padrão (σ =0,001 Å), de qualidade superior em relação aos demais. A Tabela 2 traz um conjunto de correlações para o parâmetro R2, quando tomados os valores médios de rC-C para diferentes pares de métodos. Fica claro em tal ilustração o quanto as distâncias médias fornecidas pelo método semi-empírico PM3 estão bem correlacionadas com aquelas obtidas no nível DFT, mais rigoroso no que diz respeito ao formalismo teórico, e o quanto o método HF/CEP-4G se mostrou destoante para a propriedade mencionada.
) obtido com o método semi-empírico destoa em apenas 0,01 Å em relação às distancias fornecidas pelos funcionais e mostrou parâmetros de ajuste linear, tais como o quadrado do coeficiente de correlação (R2=0,920) e desvio padrão (σ =0,001 Å), de qualidade superior em relação aos demais. A Tabela 2 traz um conjunto de correlações para o parâmetro R2, quando tomados os valores médios de rC-C para diferentes pares de métodos. Fica claro em tal ilustração o quanto as distâncias médias fornecidas pelo método semi-empírico PM3 estão bem correlacionadas com aquelas obtidas no nível DFT, mais rigoroso no que diz respeito ao formalismo teórico, e o quanto o método HF/CEP-4G se mostrou destoante para a propriedade mencionada.
Contendo a maior estrutura avaliada 13 Å de extensão e estando a convergência para os valores das distâncias médias C-C em seu início, pode-se intuir que seja plausível tomar para avaliação das demais propriedades uma estrutura a(5,5) de extensão superior a esta, pois assim, propriedades estruturais, tal como o diâmetro, já não estariam sofrendo influência tão significativa do comprimento do tubo.
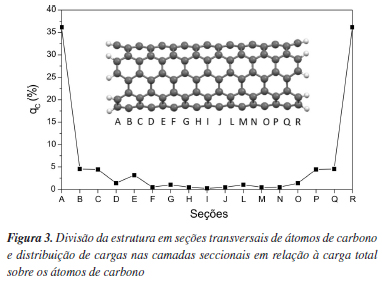
Tomando o método PM3 como um método computacionalmente barato e satisfatório para a descrição das estruturas, avaliou-se a densidade de carga sobre cada átomo, da espécie tomada para estudo, a(5,5) C170H20 (20 Å de comprimento, k = 14). A saturação das extremidades da estrutura com átomos de hidrogênio causa um efeito de borda, o que proporciona um ambiente químico diferenciado nas regiões extremas do tubo. Para avaliar esse efeito, dividiu-se a estrutura em secções transversais de átomos de carbono, cada uma contendo 10 átomos, sendo simbolizadas por letras latinas (de A a R). Devido à diferença de eletronegatividade entre os átomos de C e H, observou-se um acúmulo de carga de -2,4 u.a. sobre os átomos de C e, conseqüentemente, +2,4 u.a. de carga sobre os átomos de H, para carga total nula. A Figura 3 mostra em percentagem a distribuição de carga sobre cada secção de átomos. Pode-se observar que mais de 70% da densidade de carga negativa se encontra nas secções das extremidades A e R, diretamente ligadas aos átomos de hidrogênio. Na porção central da estrutura, de G a L, encontram-se densidades de carga mais baixas, sendo de 0,26% para a seção central I. Portanto, esta é uma região menos influenciada pelo efeito de borda, apta a receber a inserção covalente dos ligantes.
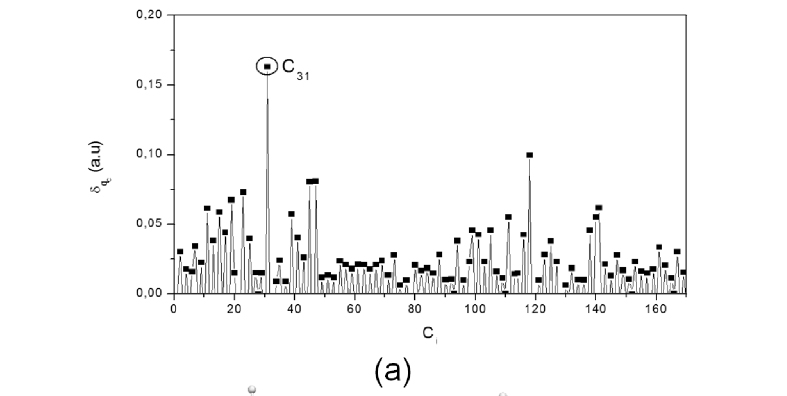
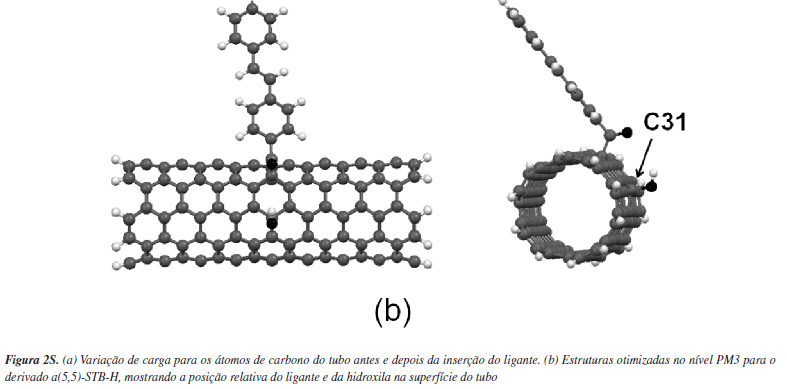
Os substituintes escolhidos são os derivados do estilbeno mostrados na Figura 1b. A ligação química de grupamentos em CNTs pode ocorrer do ponto de vista experimental, basicamente por dois caminhos: pela interação direta do grupamento a ser adicionado com um átomo de carbono presente no tubo, como na fluoração,49 ou por sua interação com grupos funcionais pré-existentes na nanoestrutura, geralmente oriundos de processos oxidativos,50 como na formação de amidas através da reação de amônia ou aminas com espécies na forma CNT-COOH.51 O modelo aqui utilizado faz referência ao segundo caminho, considerando a "pré-existência" de um grupamento carboxila no tubo, ligado em um átomo de carbono com hibridização sp3 da nanoestrutura, a exemplo do modelo utilizado por Veloso e colaboradores.52 Para que a carga do tubo fosse nula e sua multiplicidade unitária, adicionou-se também uma hidroxila em sua superfície, como é comum em CNTs oxidados. A posição de ligação da hidroxila foi definida seguindo o seguinte procedimento. A partir da inserção dos derivados de estilbeno, avaliou-se a variação da densidade de carga sobre cada átomo de carbono do tubo (δqC = qCm - qCn) modificado (qCm, carga total igual a +1 u.a.) e não modificado (qCn, carga total nula) (Figura 2S-a). Determinou-se, assim, o posicionamento da hidroxila nos átomos de menor densidade de carga, para cada sistema a(5,5)-STB-X, sendo esta posição coincidente para todos os substituintes, independente do grupamento X. Os posicionamentos dos estilbenos e de suas respectivas hidroxilas estão indicados na Figura 2S-b.
A inserção covalente do ligante (STB-X) no nanotubo de carbono provoca mudanças estruturais importantes, independentes do grupamento X, portanto, os resultados discutidos a seguir possuem um caráter geral para a série de moléculas estudadas, como pode ser observado na Tabela 3 e Figuras 4 e 3S.
Os átomos de carbono participantes das ligações com os substituintes (C2 e C7, Figura 4), antes com hibridização sp2, passam a ter características de carbono sp3, como pode ser observado através da média dos comprimentos das ligações C-C (=1,53 Å) e ângulos de ligação (=109,8º) envolvendo os átomos de carbono C1 a C8. Na Figura 4 pode-se também observar que, devido ao posicionamento dos átomos, existe uma interação fraca entre o oxigênio do grupo carbonila e o hidrogênio da hidroxila, sendo ro2-H1 = 3,3 Å. Outra característica importante é a conformação da estrutura do estilbeno em relação à carbonila. O valor do ângulo diedro δO2-C9-C10-C11 foi de 83,9º, mostrando que esses dois grupamentos são arranjados de forma perpendicular, possivelmente devido à proximidade que resultaria entre os átomos de hidrogênio do ligante e a superfície do tubo.
Com o objetivo de verificar a extensão dos defeitos estruturais na superfície do tubo, ocasionadas devido à presença do ligante, avaliaram-se nas direções longitudinal (Figura 3S-a) e transversal (Figura 3S-b) as alterações nas distâncias C-C, em relação à estrutura do tubo isolado (δr). A Figura 3S-a mostra claramente uma significativa alteração nas distâncias de ligação envolvendo os átomos ligados ao carbono no qual está inserido o derivado do estilbeno (ligações H e I), no entanto, na direção longitudinal, o defeito se estende apenas por quatro ligações C-C (de G a J na Figura 3S-a). Pode-se observar também na Figura 3S-b, que o defeito na direção transversal é mais significativo nas proximidades dos átomos de carbono ligados ao derivado do estilbeno (ligações h a m). Este efeito mostrou-se também localizado nas regiões próximas aos ligantes.
Propriedades ópticas não-lineares
Os cálculos dos elementos do tensor da primeira hiperpolarizabilidade (βijk) foram efetuados utilizando os métodos CPHF e TDHF como implementado nos pacotes computacionais Gaussian-03 e MOPAC-07, respectivamente. Os valores de <β> estão contidos na Tabela 4, sendo obtidos para os substituintes livres (Y-STB-X, Y=COOH) e para os tubos modificados (a(5,5)-STB-X). Pode-se observar na Tabela 4 que os cálculos em ambos os níveis de teoria atribuíram valores baixos para o <β> do tubo isolado (<4,5x10-30 cm5 esu-1), consistente com a alta simetria do sistema (D5h). Ambos os métodos mostraram-se destoantes do ponto de vista quantitativo, apresentando, de forma geral, tendências equivalentes.
Na Figura 5a os resultados DFT são representados na forma de gráficos de dispersão, com o substituinte X descrito pelo parâmetro eletrônico de Hammett (σp).53 Os valores de σp foram definidos por Hammett a partir das constantes de ionização do ácido benzóico, como mostrado na Equação 4


sendo KH a constante de ionização em água a 25 ºC para o ácido benzóico e KX a correspondente constante para o derivado substituído na posição para. O descritor σp pode ser interpretado como o efeito polar (eletrônico) exercido pelo substituinte X. Grupos doadores de elétrons possuem σp < 0 e grupos aceptores de elétrons σp > 0, sendo a magnitude do valor absoluto uma medida da extensão do efeito eletrônico. Para os sistemas considerados no presente estudo, σp apresenta valores na faixa de -0,66 (X=NH2, doador de elétrons forte) a 0,78 (X=NO2, aceptor de elétrons forte).
Os derivados do estilbeno 4,4´-substituídos são reconhecidos como compostos importantes no desenho de materiais com propriedades NLO.16,54,55 Para esses sistemas di-aromáticos, é necessário que os substituintes nas posições 4 e 4´ apresentem efeitos eletrônicos em sentidos opostos, isto é, grupos doadores e retirados de elétrons devem estar presentes gerando moléculas do tipo push-pull. Os derivados representados na Figura 1b contêm o grupo COR em uma das extremidades, o qual possui características aceptoras de elétrons independente do grupo R (σp~0,5, ver ref. 53). Portanto, é esperado um aumento de <β> para análogos em que o substituinte X é um doador de elétrons (σp<0). Esse comportamento pode ser observado na Figura 5a para o nível de teoria DFT-SVWN/6-31G(d)//PM3. No nível DFT, <β> (em unidades de 10-30 cm5 esu-1) varia de 37,7 (X=H) a 110,1 (X=NH2) para os derivados de estilbeno livre e de 54,1 (X=H) a 187,5 (X=NH2) para os nanotubos modificados. Apesar da menor sensibilidade dos valores de <β> no nível PM3 (Tabela 4), o comportamento qualitativo é equivalente aos resultados DFT, mostrando um crescimento linear da propriedade com o poder doador de elétrons do substituinte X (ver valores para os substituintes X=H, OH e NH2). No nível DFT, com exceção do derivado com X=H (σp=0), a variação linear de <β> pode ser estendida para grupos aceptores de elétrons, com <β> diminuindo na ordem NH2 > OH > Cl > NO2 (Figura 5a).
Além do efeito do substituinte X no valor de <β> dos compostos analisados, é importante ressaltar as mudanças na propriedade em conseqüência da inserção dos derivados de estilbeno na superfície do nanotubo de carbono. Analisando-se os valores da Tabela 4 podem ser observadas tendências distintas em função do nível de teoria. No nível PM3 os valores de <β> diminuem para os nanotubos modificados e no nível DFT a propriedade é intensificada em relação ao derivado do estilbeno livre correspondente (Figura 5a). Os análogos com X=NO2 desviam desse comportamento geral em ambos os níveis de teoria. É interessante notar que no nível PM3 a diminuição nos valores de <β> é praticamente constante para X=Cl, H, OH e NH2, estando na faixa de 8-13x10-30 cm5 esu-1. Entretanto, no nível DFT-SVWN/6-31G(d)//PM3 a taxa de crescimento de <β> é proporcional ao efeito doador de elétrons do substituinte X, variando de 6x10-30 (X=Cl, σp=0,23) a 78x10-30 cm5 esu-1 (X=NH2, σp=-0,66). Em última análise, a dependência de <β> com a freqüência do campo aplicado foi avaliada no nível PM3. Os resultados são apresentados na Tabela 4 para a energia do campo igual a 0,65 eV. Como se pode observar, as variações são pequenas nesse nível de teoria, inferiores a 30%, sendo o comportamento qualitativo similar àquele obtido para a energia nula do campo. É importante realçar as limitações do nível PM3, as quais não permitem uma análise quantitativa e definitiva do efeito do campo externo nos valores da primeira hiperpolarizabilidade. Entretanto, do ponto de vista qualitativo, os resultados mostraram que os valores de <β> calculados para ħω = 0,00eV, em ambos os níveis de teoria, devem ser inferiores aos valores observados, colocando os sistemas estudados como potenciais agentes para aplicação em óptica não linear.
Finalmente, a Figura 5b ilustra os resultados discutidos anteriormente para o nível DFT, evidenciando o efeito do substituinte X e do nanotubo de carbono nos valores de <β>, dentro da aproximação estática. Para os tubos modificados com derivados do estilbeno contendo grupos doadores de elétrons (X=NH2) na posição 4, os valores de <β> são expressivos dentro do contexto de sistemas com potencial em NLO (<β> =187,5x10-30 cm5 esu-1 para a(5,5)-STB-NH2). Como exemplo, esquaraínas não simétricas, sintetizadas por Chen e colaboradores,55 apresentaram valores da primeira hiperpolarizabilidade estática de 170x10-30 cm5 esu-1. Esses compostos têm sido considerados candidatos atrativos para o desenvolvimento de novos materiais com diversas aplicações em NLO. Portanto, a proposta discutida neste trabalho e os resultados de relação entre estrutura e propriedade podem ser considerados como evidências no nível molecular para o planejamento de novos nanotubos de carbono quimicamente funcionalizados. Vale mencionar que os resultados apresentados foram obtidos para uma estrutura com diâmetro da ordem de 0,7 nm e comprimento de 20 Å. Propriedades ópticas de CNTs dependem da dimensão da estrutura, como descrito recentemente por Jensen e colaboradores.19 Nesse trabalho, os autores avaliaram o efeito do comprimento, diâmetro e quiralidade de CNTs na polarizabilidade (α) e segunda hiperpolarizabilidade (γ). Os resultados levaram a conclusões relevantes, como a existência de um comprimento de saturação, dependente, principalmente, da propriedade (~45 Å para α e ~75 Å para γ); a intensificação dos valores com o aumento do diâmetro para a série a(5,5)-a(10,10) e, uma pequena diminuição dos valores calculados de γ com o fechamento das extremidades dos tubos. Não há na literatura um estudo detalhado para o comportamento de β com as propriedades estruturais de CNTs, no entanto, esperam-se tendências intermediárias entre os resultados para α e γ, ou seja, distâncias de saturação entre 40 e 70 Å e intensificação da grandeza com o aumento do diâmetro. Outros estudos estão em andamento no nosso grupo, visando ampliar a análise, incluindo a dependência da propriedade com outros aspectos estruturais dos nanotubos de carbono, como diâmetro, quiralidade, presença de defeitos estruturais e múltiplas camadas. Esses resultados serão relatados em trabalhos futuros.
CONCLUSÕES
Neste trabalho, métodos quânticos foram utilizados para a análise da estrutura e propriedades ópticas não lineares (NLO) de nanotubos de carbono (CNTs) quimicamente modificados. O tubo armchair a(5,5) foi escolhido como suporte para a inserção covalente de derivados do estilbeno (STB-X) contendo substituintes doadores e aceptores de elétrons (X=NO2, Cl, H, OH, NH2).
A análise da adequação da metodologia para a representação da estrutura desses sistemas mostrou que o método semi-empírico PM3 fornece resultados equivalentes àqueles obtidos nos níveis DFT PBE e B3LYP/6-31G(d). Considerando a distância média rC-C extrapolada para o comprimento infinito do tubo, os valores obtidos foram 1,42 (PM3) e 1,43 Å (DFT), sendo o valor esperado igual a 1,44 Å. Portanto, pode-se concluir que o método semi-empírico PM3 fornece estruturas satisfatórias para CNTs de parede única e, portanto, representa uma alternativa com custo computacional moderado para o estudo de sistemas com dimensões nanométricas.
As geometrias dos CNTs modificados foram obtidas no nível PM3 e a primeira hiperpolarizabilidade estática (<β>) calculada com os formalismos CPHF (SVWN/6-31G(d)//PM3) e TDHF (PM3). Ambos os métodos fornecem tendências equivalentes para a série de moléculas estudadas, entretanto, os valores DFT apresentaram maiores magnitudes. No nível SVWN/6-31G(d)//PM3, observou-se um aumento de <β> com o poder doador de elétrons do substituinte X, medido através do descritor eletrônico de Hammett (σp). Os valores de <β> (em 10-30 cm5 esu-1) variaram de 27,2 (X=NO2) a 187,5 (X=NH2), sendo observado um aumento da propriedade de até 70% (para X=NH2) quando o derivado do estilbeno é inserido na superfície do tubo. Portanto, os resultados obtidos permitem estabelecer uma relação entre estrutura e propriedade, a qual pode ser definida em duas partes: a inserção do substituinte (STB-X) na superfície do tubo intensifica o valor de <β>, sendo o aumento dependente de X e, o valor de <β> para o tubo modificado (a(5,5)-STB-X) aumenta de forma linear com o poder doador de elétrons do substituinte X, medido através do parâmetro eletrônico de Hammett (σp).
Finalmente, os resultados e conclusões obtidos neste trabalho colocam em evidência características interessantes de nanotubos de carbono modificados, as quais devem ser consideradas e exploradas em estudos futuros para sistemas análogos e modelos mais representativos da realidade.
MATERIAL SUPLEMENTAR
O material suplementar, disponível em http://quimicanova.sbq.org.br, em arquivo pdf e com acesso livre, traz figuras que ilustram a variação da distância (rC-C) em função do número de átomos de carbono, obtida no nível PM3 (Figura 1S); a variação de carga em cada átomo de carbono com a inserção do ligante cromóforo (Figura 2S) e avaliação do efeito estrutural causado no tubo devido ao ligante (Figura 3S).
AGRADECIMENTOS
Ao CNPq e FAPEMIG (CEX-1861/06) pelo suporte financeiro. A. M. Da Silva, Jr. agradece à CAPES pela bolsa de pós-graduação. Esse trabalho foi parcialmente financiado pelo projeto PRONEX-FAPEMIG/EDT-526/07.
REFERÊNCIAS
1. Iijima. S.; Nature 1991, 354, 56.
2. Coluci, V. R.; Dantas, S. O.; Jorio, A.; Galvão, D. S.; Phys. Rev. B 2007, 75, 075417.
3. Dinadayalane, T. C.; Kaczmarek, A.; Łukaszewicz, J.; Leszczynski, J.; J. Phys. Chem. C 2007, 111, 7376.
4. Trzaskowski, B.; Jalbout, A. F.; Adamowicz, L.; Chem. Phys. Lett. 2007, 444, 314.
5. Vieira, H. S.; Andrada, D. M.; Mendonça, R.; Santos, A. P.; Martins, M. D.; Macedo, W. A. A.; Gorgulho, H. F.; Pimenta, L. P. S.; Moreira, R. L.; Jorio, A.; Pimenta, M. A.; Furtado, C. A.; J. Nanosci. Nanotechnol. 2007, 7, 3421.
6. Ramesh, S.; Shan, H.; Haroz, E.; Billups, W. E.; Hauge, R.; Adams, W. W.; Smalley, R. E.; J. Phys. Chem. C 2007, 111, 17827.
7. Feazell R. P.; Nakayama-Ratchford, N.; Daí, H.; Lippard, S. J.; J. Am. Chem. Soc. 2007, 129, 8438.
8. Ajayan, P. M.; Chem. Rev. 1999, 99, 1787.
9. Thiên-Nga, L.; Bonard, J.-M.; Gáal, R.; Forró, L.; Appl. Phys. Lett. 2002, 80, 850.
10. Dresselhaus, M. S.; Dresselhaus, G.; Saito, R.; Carbon 1995, 33, 883.
11. Anantram, M. P.; Leonard, F.; Rep. Prog. Phys. 2006, 69, 507.
12. Banerju, S.; Hemraj-Benny, T.; Wong, S. S.; Adv. Mater. 2005, 1, 17.
13. Souza-Filho, A. G.; Fagan, S. B.; Quim. Nova 2007, 30, 1695.
14. Mateo-Alonso, A.; Ehli, C.; Chen, K. H.; Guldi, D. M.; Prato, M.; J. Phys. Chem. A 2007, 111, 12669.
15. Kanis, D. R.; Ratner, M. A.; Marks, T. J.; Chem. Rev. 1994, 94. 195.
16. Cheng, L.-T.; Tam, W.; Stevenson, S. H.; Meredith, G. R.; Rikken, G.; Marder, S. R.; J. Phys. Chem. 1991, 95, 10631.
17. Xie, R.-H.; J. Chem. Phys. 1998, 108, 3626.
18. Jensen, L.; Schmidt, O. H.; Mikkelsen, K. V.; Åstrand, P.-O.; J. Phys. Chem. B 2000, 104, 10462.
19. Jensen, L.; Åstrand, P.-O.; Mikkelsen, K. V.; J. Phys. Chem. A 2004, 108, 8795.
20. De Dominicis, L.; Botti, S.; Asilyan, L. S.; Ciardi, R.; Fantoni, R.; Terranova, M. L.; Fiori, A.; Orlanducci, S.; Appolloni, R.; Appl. Phys. Lett. 2004, 85, 1418.
21. Guo, G. Y.; Chu, K. C.; Wang, D.-S.; Duan, C.-G.; Phys. Rev. B 2004, 69, 205416.
22. Sun, J.; Guo, Z.; Liang, W.; Phys. Rev. B 2007, 75, 195438.
23. Ding, F.; Phys. Rev. B 2005, 72, 245409.
24. Tapas, K.; Brahin, A.; Xiaofeng, D.; Pachter, R.; Chem. Phys. Lett. 2004, 392, 176.
25. Wanbayor, R.; Ruangpornvisuti, V.; Chem. Phys. Lett. 2007, 441, 127.
26. Petsalakis, I. D.; Pagona, G.; Tagmatarchis, N.; Theodorakopoulos, G.; Chem. Phys. Lett. 2007, 448, 115.
27. Stewart, J. J. P.; J. Comput. Chem. 1989, 10, 221.
28. Szaboo, A.; Ostlund, N. S.; Modern Quantum Chemistry: Introduction to advanced eletronic structure theory, Dove Publications: New York, 1996.
29. Cundari, T. R.; Stevens, W. J.; J. Chem. Phys. 1993, 98, 5555.
30. Collins, J. B.; Schleyer, P. V. R.; Binkley, J. S.; Pople, J. A.; J. Chem. Phys. 1976, 64, 5142.
31. Gordon, M. S.; Binkley, J. S.; Pople, J. A.; Pietro, W. J.; Hehre, W. J.; J. Am. Chem. Soc. 1982, 104, 2797.
32. Duarte, H. A.; Quim. Nova 2001, 24, 501.
33. Perdew, J. P.; Burke, K.; Ernzerhof, M.; Phys. Rev. Lett. 1997, 78, 1396.
34. Becke, A. D.; Phys. Rev. A 1988, 38, 3098; Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785.
35. Feynman, R.; Phys. Rev. 1939, 56, 340; Stanton, R. E.; J. Chem. Phys. 1962, 36, 1298; Pople, J. A.; McIver, J. M.; Ostlund, N. S.; J. Chem. Phys. 1968, 49, 2960.
36. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03 Revision D.02, Gaussian, Inc., Wallingford CT, 2004.
37. Kurtz, H. A.; Stewart, J. J. P.; Dieter, K. M.; J. Comput. Chem. 1990, 11, 82; Dupuis, M.; Karna, S. P.; J. Comput. Chem. 1991, 12, 487.
38. MOPAC2007;- James, J. P.; Stewart Computational Chemistry, Colorado Springs, CO, USA, http://openmopac.net (2007).
39. Champagne, B.; Perpète, E. A.; Jacquemin, D.; van Gisbergen, S. J. A.; Baerends, E.-J.; Soubra-Ghaoui, C.; Robins, K. A.; Kirtman, B.; J. Phys. Chem. A 2000, 104, 4755.
40. Yang, G. C.; Shi, S. Q.; Guan, W.; Fang, L.; Su, Z. M.; J. Mol. Struct. (Theochem) 2006, 773, 9.
41. Davidson, E. R.; Eichinger, B. E.; Robinson, B. H.; Optical Mat. 2006, 29, 360.
42. Isborn, C. M.; Leclercq A.; Vila, F. D.; Dalton, L. R.; Brédas, J. L.; Eichinger, B. E.; Robinson, B. H.; J. Phys. Chem. A 2007, 111, 1319.
43. Ferreira, D. E. C.; Dos Santos, H. F.; De Almeida, W. B.; Junqueira, G. M. A.; J. Braz. Chem. Soc. 2007, 18, 1379.
44. Kleinman, D. A.; Phys. Rev. 1977, 126, 1962.
45. Nandi, P. K.; Mandal, K.; Kar, T.; J. Mol. Struct.: THEOCHEM 2006, 760, 235.
46. Bettinger, H. F.; Org. Lett. 2004, 6, 731.
47. Saito, R.; Dresselhaus, G.; Dresselhaus, M. S.; Physical Properties of Carbon Nanotubes, Imperial College: London, 1998.
48. Barros, E. M.; Jorio, A.; Samsonidze, G. G.; Capaz, R. B.; Souza-Filho, A. G.; Mendes-Filho, J.; Dresselhaus, G.; Dresselhaus, M. S.; Phys. Rep. 2006, 431, 261.
49. Touhara, H.; Okino, F.; Carbon 2000, 38, 241.
50. Zhang, J.; Zou, H.; Qing, Q.; Yang, Y.; Li, Q.; Liu, Z.; Guo, X.; Du, Z.; J. Phys. Chem. B 2003, 107, 3712.
51. Banerjee, S.; Hemraj-Benny, T.; Wong, S. S.; Adv. Mater. 2005, 17, 17.
52. Veloso, M. V.; Souza-Filho, A. G.; Mendes-Filho, J.; Fagan, S. B.; Mota, R.; Chem. Phys. Lett. 2006, 430, 71.
53. Hansch, C.; Leo, A.; Taft, R. W.; Chem. Rev. 1991, 91, 165.
54. Amatatsu, Y.; Ohara, Y.; Chem. Phys. Lett. 2003, 373, 245.
55. Chen, C.-T.; Marder, S. R.; Cheng, L.-T.; J. Am. Chem. Soc. 1994, 116, 3117.
Recebido em 29/1/08; aceito em 10/9/08; publicado na web em 2/2/09
Material Suplementar

Figura 1S - clique para ampliar

Figura 2S - clique para ampliar

Figura 3S - clique para ampliar
- 1. Iijima. S.; Nature 1991, 354, 56.
- 2. Coluci, V. R.; Dantas, S. O.; Jorio, A.; Galvão, D. S.; Phys. Rev. B 2007, 75, 075417.
- 3. Dinadayalane, T. C.; Kaczmarek, A.; Łukaszewicz, J.; Leszczynski, J.; J. Phys. Chem. C 2007, 111, 7376.
- 4. Trzaskowski, B.; Jalbout, A. F.; Adamowicz, L.; Chem. Phys. Lett 2007, 444, 314.
- 5. Vieira, H. S.; Andrada, D. M.; Mendonça, R.; Santos, A. P.; Martins, M. D.; Macedo, W. A. A.; Gorgulho, H. F.; Pimenta, L. P. S.; Moreira, R. L.; Jorio, A.; Pimenta, M. A.; Furtado, C. A.; J. Nanosci. Nanotechnol 2007, 7, 3421.
- 6. Ramesh, S.; Shan, H.; Haroz, E.; Billups, W. E.; Hauge, R.; Adams, W. W.; Smalley, R. E.; J. Phys. Chem. C 2007, 111, 17827.
- 7. Feazell R. P.; Nakayama-Ratchford, N.; Daí, H.; Lippard, S. J.; J. Am. Chem. Soc 2007, 129, 8438.
- 8. Ajayan, P. M.; Chem. Rev 1999, 99, 1787.
- 9. Thiên-Nga, L.; Bonard, J.-M.; Gáal, R.; Forró, L.; Appl. Phys. Lett 2002, 80, 850.
- 10. Dresselhaus, M. S.; Dresselhaus, G.; Saito, R.; Carbon 1995, 33, 883.
- 11. Anantram, M. P.; Leonard, F.; Rep. Prog. Phys 2006, 69, 507.
- 12. Banerju, S.; Hemraj-Benny, T.; Wong, S. S.; Adv. Mater 2005, 1, 17.
- 13. Souza-Filho, A. G.; Fagan, S. B.; Quim. Nova 2007, 30, 1695.
- 14. Mateo-Alonso, A.; Ehli, C.; Chen, K. H.; Guldi, D. M.; Prato, M.; J. Phys. Chem. A 2007, 111, 12669.
- 15. Kanis, D. R.; Ratner, M. A.; Marks, T. J.; Chem. Rev 1994, 94 195.
- 16. Cheng, L.-T.; Tam, W.; Stevenson, S. H.; Meredith, G. R.; Rikken, G.; Marder, S. R.; J. Phys. Chem 1991, 95, 10631.
- 17. Xie, R.-H.; J. Chem. Phys 1998, 108, 3626.
- 18. Jensen, L.; Schmidt, O. H.; Mikkelsen, K. V.; Åstrand, P.-O.; J. Phys. Chem. B 2000, 104, 10462.
- 19. Jensen, L.; Åstrand, P.-O.; Mikkelsen, K. V.; J. Phys. Chem. A 2004, 108, 8795.
- 20. De Dominicis, L.; Botti, S.; Asilyan, L. S.; Ciardi, R.; Fantoni, R.; Terranova, M. L.; Fiori, A.; Orlanducci, S.; Appolloni, R.; Appl. Phys. Lett 2004, 85, 1418.
- 21. Guo, G. Y.; Chu, K. C.; Wang, D.-S.; Duan, C.-G.; Phys. Rev. B 2004, 69, 205416.
- 22. Sun, J.; Guo, Z.; Liang, W.; Phys. Rev. B 2007, 75, 195438.
- 23. Ding, F.; Phys. Rev. B 2005, 72, 245409.
- 24. Tapas, K.; Brahin, A.; Xiaofeng, D.; Pachter, R.; Chem. Phys. Lett 2004, 392, 176.
- 25. Wanbayor, R.; Ruangpornvisuti, V.; Chem. Phys. Lett 2007, 441, 127.
- 26. Petsalakis, I. D.; Pagona, G.; Tagmatarchis, N.; Theodorakopoulos, G.; Chem. Phys. Lett 2007, 448, 115.
- 27. Stewart, J. J. P.; J. Comput. Chem 1989, 10, 221.
- 28. Szaboo, A.; Ostlund, N. S.; Modern Quantum Chemistry: Introduction to advanced eletronic structure theory, Dove Publications: New York, 1996.
- 29. Cundari, T. R.; Stevens, W. J.; J. Chem. Phys 1993, 98, 5555.
- 30. Collins, J. B.; Schleyer, P. V. R.; Binkley, J. S.; Pople, J. A.; J. Chem. Phys 1976, 64, 5142.
- 31. Gordon, M. S.; Binkley, J. S.; Pople, J. A.; Pietro, W. J.; Hehre, W. J.; J. Am. Chem. Soc 1982, 104, 2797.
- 32. Duarte, H. A.; Quim. Nova 2001, 24, 501.
- 33. Perdew, J. P.; Burke, K.; Ernzerhof, M.; Phys. Rev. Lett 1997, 78, 1396.
- 34. Becke, A. D.; Phys. Rev. A 1988, 38, 3098;
- Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785.
- 35. Feynman, R.; Phys. Rev 1939, 56, 340;
- Stanton, R. E.; J. Chem. Phys 1962, 36, 1298;
- Pople, J. A.; McIver, J. M.; Ostlund, N. S.; J. Chem. Phys 1968, 49, 2960.
- 36. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03 Revision D.02, Gaussian, Inc., Wallingford CT, 2004.
- 37. Kurtz, H. A.; Stewart, J. J. P.; Dieter, K. M.; J. Comput. Chem 1990, 11, 82; Dupuis, M.; Karna, S. P.; J. Comput. Chem 1991, 12, 487.
- 38. MOPAC2007;- James, J. P.; Stewart Computational Chemistry, Colorado Springs, CO, USA, http://openmopac.net (2007).
- 39. Champagne, B.; Perpète, E. A.; Jacquemin, D.; van Gisbergen, S. J. A.; Baerends, E.-J.; Soubra-Ghaoui, C.; Robins, K. A.; Kirtman, B.; J. Phys. Chem. A 2000, 104, 4755.
- 40. Yang, G. C.; Shi, S. Q.; Guan, W.; Fang, L.; Su, Z. M.; J. Mol. Struct. (Theochem) 2006, 773, 9.
- 41. Davidson, E. R.; Eichinger, B. E.; Robinson, B. H.; Optical Mat 2006, 29, 360.
- 42. Isborn, C. M.; Leclercq A.; Vila, F. D.; Dalton, L. R.; Brédas, J. L.; Eichinger, B. E.; Robinson, B. H.; J. Phys. Chem. A 2007, 111, 1319.
- 43. Ferreira, D. E. C.; Dos Santos, H. F.; De Almeida, W. B.; Junqueira, G. M. A.; J. Braz. Chem. Soc 2007, 18, 1379.
- 44. Kleinman, D. A.; Phys. Rev 1977, 126, 1962.
- 45. Nandi, P. K.; Mandal, K.; Kar, T.; J. Mol. Struct.: THEOCHEM 2006, 760, 235.
- 46. Bettinger, H. F.; Org. Lett 2004, 6, 731.
- 47. Saito, R.; Dresselhaus, G.; Dresselhaus, M. S.; Physical Properties of Carbon Nanotubes, Imperial College: London, 1998.
- 48. Barros, E. M.; Jorio, A.; Samsonidze, G. G.; Capaz, R. B.; Souza-Filho, A. G.; Mendes-Filho, J.; Dresselhaus, G.; Dresselhaus, M. S.; Phys. Rep 2006, 431, 261.
- 49. Touhara, H.; Okino, F.; Carbon 2000, 38, 241.
- 50. Zhang, J.; Zou, H.; Qing, Q.; Yang, Y.; Li, Q.; Liu, Z.; Guo, X.; Du, Z.; J. Phys. Chem. B 2003, 107, 3712.
- 51. Banerjee, S.; Hemraj-Benny, T.; Wong, S. S.; Adv. Mater 2005, 17, 17.
- 52. Veloso, M. V.; Souza-Filho, A. G.; Mendes-Filho, J.; Fagan, S. B.; Mota, R.; Chem. Phys. Lett 2006, 430, 71.
- 53. Hansch, C.; Leo, A.; Taft, R. W.; Chem. Rev 1991, 91, 165.
- 54. Amatatsu, Y.; Ohara, Y.; Chem. Phys. Lett 2003, 373, 245.
- 55. Chen, C.-T.; Marder, S. R.; Cheng, L.-T.; J. Am. Chem. Soc 1994, 116, 3117.
Datas de Publicação
-
Publicação nesta coleção
23 Abr 2009 -
Data do Fascículo
2009
Histórico
-
Recebido
29 Jan 2008 -
Aceito
10 Set 2008