Resumo
Methodologies of extraction of lipids from chicken breast and oats flakes were evaluated: Soxhlet, Folch et al., Bligh & Dyer and Hara & Radin. For chicken breast, the methods Soxhlet, Folch et al. and Bligh & Dyer presented the highest yields in total lipids. With oat flakes, the methods Soxhlet and Bligh & Dyer presented higher yields than the Hara & Radin and Folch et al. The Soxhlet method affected the quality of the lipid fraction in both samples. Extracted lipid components were separated by thin layer chromatography, the chloroform-methanol based was more efficient to extract the neutral and polar lipids.
lipids; methods of extraction; lipids quality
lipids; methods of extraction; lipids quality
ARTIGO
Métodos de extração e qualidade da fração lipídica de matérias-primas de origem vegetal e animal
Extraction methods and quality of the lipid fraction of vegetable and animal samples
Aelson Aloir Santana Brum; Lia Ferraz de Arruda; Marisa Aparecida Bismara Regitano-d´Arce* * e-mail: mabra@esalq.usp.br
Departamento de Agroindústria, Alimentos e Nutrição, Escola Superior de Agricultura Luiz de Queiroz, Universidade de São Paulo, CP 9, 13418-900 Piracicaba - SP, Brasil
ABSTRACT
Methodologies of extraction of lipids from chicken breast and oats flakes were evaluated: Soxhlet, Folch et al., Bligh & Dyer and Hara & Radin. For chicken breast, the methods Soxhlet, Folch et al. and Bligh & Dyer presented the highest yields in total lipids. With oat flakes, the methods Soxhlet and Bligh & Dyer presented higher yields than the Hara & Radin and Folch et al. The Soxhlet method affected the quality of the lipid fraction in both samples. Extracted lipid components were separated by thin layer chromatography, the chloroform-methanol based was more efficient to extract the neutral and polar lipids.
Keywords: lipids; methods of extraction; lipids quality.
INTRODUÇÃO
A extração de óleo com solvente é um processo de transferência de constituintes solúveis (o óleo) de um material inerte (a matriz graxa) para um solvente com o qual a matriz se acha em contato. Os processos que ocorrem são meramente físicos, pois o óleo transferido para o solvente é recuperado sem nenhuma reação química.1
A extração de lipídios é uma determinação importante em estudos bioquímicos, fisiológicos e nutricionais dos mais diversos tipos de alimentos e, portanto, deve ser realizada com acurácia. Algumas amostras requerem cuidados especiais para a obtenção da fração lipídica, pois fatores como co-extração dos componentes não-lipídicos e a oxidação indesejada podem influenciar a qualidade final da fração lipídica. Os procedimentos clássicos idealizados por Soxhlet em 1879, com refluxo de solvente por muitas horas, devem ser evitados, já que favorecem as reações de peroxidação e de hidrólise,2 podendo comprometer resultados analíticos posteriores, como a quantificação de certos componentes lipídicos. Um dos procedimentos de extração mais versáteis e efetivos, que supera as dificuldades mencionadas acima, é a metodologia de Bligh & Dyer,3 uma versão simplificada do procedimento clássico usando clorofórmio-metanol proposto por Folch et al..4
Outra vantagem apresentada pelos métodos baseados na mistura binária clorofórmio e metanol é a capacidade de extraírem tanto os lipídios neutros e os lipídios polares eficientemente. Diversos estudos mostraram que, dependendo do tipo de tecido que será analisado, a escolha do método de extração influencia significantemente no resultado final.2,5-7
A insolubilidade dos lipídios em água torna possível sua separação das proteínas, carboidratos e da água nos tecidos. Como os lipídios têm uma grande faixa de relativa hidrofobicidade, é praticamente inviável a utilização de um único solvente universal para a extração dos lipídios.5 Lipídios neutros estão ligados covalentemente e podem ser extraídos dos tecidos por solventes apolares, enquanto lipídios polares, os quais estão ligados por forças eletrostáticas e pontes de hidrogênio, requerem solventes polares capazes de quebrar tais ligações e liberá-los.
O primeiro aparelho para extração dos lipídios em matrizes graxas foi desenvolvido por Franz von Soxhlet em 1879,8 que ressaltou a importância do grau de trituração da amostra quanto à duração e eficácia do processo. No processo de liberação extrativa, levam-se em conta três etapas principais: a penetração do solvente no tecido; a formação de uma miscela intracelular e, a difusão do extrato na miscela externa.9 Consiste no tratamento sucessivo e intermitente da amostra imersa em um solvente puro (éter de petróleo, éter dietílico ou n-hexano), graças à sifonagem e subseqüente condensação do solvente aquecido dentro do balão que está na base do aparelho.8
As mais notáveis vantagens que o método de Soxhlet apresenta são a amostra está sempre em contato com o solvente, havendo sua constante renovação; a temperatura do sistema mantém-se relativamente alta, visto que o calor aplicado para o processo de evaporação é constante; é uma metodologia muito simples que não requer treinamento especializado e que possibilita a extração de uma quantidade maior de óleo em relação a outros métodos, sem a necessidade de filtração da miscela após o término da extração, pois a amostra esteve envolta no cartucho durante todo o procedimento.10
O uso de um único solvente não é recomendável para a extração dos lipídios de tecidos animais. A mistura de solventes ideal para extração da matéria graxa de tecidos deve ser suficientemente polar para removê-la das associações com as membranas celulares ou com lipoproteínas, sem que ocorra reação química.6
Pesquisadores, como Folch et al.,4 reconheceram estas características intrínsecas da extração de lipídios e desenvolveram um método usando uma mistura de clorofórmio e metanol, seguida pela adição de solução de cloreto de potássio, visando uma melhor separação das fases. Bligh e Dyer3 modificaram o método de Folch et al.4 e propuseram um "método rápido para extração e purificação dos lipídios totais".
Apesar de algumas desvantagens, como a toxidez dos solventes usados e a indesejável extração dos contaminantes não-lipídicos da fase orgânica, os métodos de Folch et al.4 e Bligh e Dyer3 são largamente utilizados, tanto como propostos originalmente, quanto modificados.6,11-18
Uma das vantagens do método desenvolvido por Bligh e Dyer3 é a formação de um sistema bifásico a partir das proporções de solventes adicionados durante o processo de extração. A formação desse sistema bifásico está baseada na teoria do equilíbrio líquido-líquido de três componentes (clorofórmio/metanol/água). A determinação das solubilidades de cada componente pode ser avaliada através de um diagrama ternário de solubilidade de dois líquidos parcialmente miscíveis entre si (clorofórmio e água) com um terceiro (metanol), completamente miscível nos outros dois.
Nos últimos anos diversos pesquisadores realizaram trabalhos16,17,19-23 em que o objetivo foi encontrar o método mais eficiente para a obtenção da fração lipídica desejada e determinar sua composição. Além disso, também buscaram métodos que não alterassem a qualidade do óleo e utilizassem solventes menos tóxicos que a mistura clorofórmio e metanol.3,4,12 No entanto, devido à alta eficiência que este sistema apresenta na extração de lipídios polares e apolares, nenhuma mistura alternativa testada recentemente tem obtido o êxito esperado, ou seja, uma eficiência equivalente ao sistema que utiliza um solvente clorado.
Em um trabalho realizado,18 verificou-se que o método de Soxhlet, usando diferentes solventes como éter de petróleo, metanol, n-hexano, diclorometano:hexano (1:4) e acetona:hexano (1:4), separadamente em cada extração, foi conveniente para extração da fração lipídica de amostras sólidas que continham alto teor de lipídios, tais como chocolate em pó, margarina, leite em pó e ração para frango. Os métodos de Bligh e Dyer,3 usando clorofórmio-metanol-água, e Bligh e Dyer, modificado,15 utilizando isopropanol-ciclo-hexano-água, foram mais adequados para extração de amostras líquidas, tais como leite, ovos e plasma humano. As amostras extraídas com metanol, tanto por Soxhlet como por Bligh e Dyer,3 apresentaram um alto rendimento em óleo, já que o metanol possui um poder de solvatação superior ao dos outros solventes usados, extraindo os lipídios e os não-lipídios da matriz em estudo.
Gandhi et al.24 buscaram solventes alternativos para extração de óleo de soja, haja vista que o n-hexano é o solvente universal usado para extração dessa oleaginosa. Foram utilizados os solventes puros n-hexano, n-heptano, n-propanol, isopropanol e etanol, além da aplicação de sistemas de solventes aquosos (90:10 n-propanol) e azeótropos como, por exemplo, n-hexano + isopropanol (78+22), em períodos de extração crescentes até 10 h. Os pesquisadores concluíram que n-heptano, n-propanol, isopropanol e etanol foram igualmente efetivos na extração, quando comparados com o n-hexano. As misturas de solventes aquosos também foram eficientes com a vantagem de requerer um menor volume de solvente, porém com o inconveniente de demandar maior energia para remoção da água do solvente.
O objetivo deste trabalho foi comparar quatro diferentes métodos de extração de lipídios em um produto de origem animal (peito de frango) e outro de origem vegetal (aveia em flocos), identificando a composição lipídica dos extratos obtidos dos diferentes métodos aplicados e avaliando o tempo de extração, o rendimento, a toxidez dos solventes utilizados, a repetibilidade de cada método e a qualidade oxidativa da fração lipídica obtida.
PARTE EXPERIMENTAL
Matérias-primas
Foram utilizadas amostras comerciais de aveia em flocos e peito de frangos que após o abate foram embalados em sacos plásticos do tipo zip-loc e armazenados à temperatura de -18 ºC, até sua utilização nas extrações.
As amostras de aveia em flocos foram trituradas em micro moinho Tecnal do tipo Willie (TE - 048). A granulometria média (66,7% retido) da amostra triturada foi de 0,595 mm, verificada em jogo de peneiras Granutest nº 30. As amostras de peito de frango foram previamente descongeladas em refrigerador e trituradas em aparelho elétrico da marca Vicris, modelo 5068-85, contendo disco com furos de 5 mm de diâmetro.
A composição bromatológica das amostras estudadas foi realizada conforme metodologia AOAC.25
Métodos de extração da fração lipídica nas amostras selecionadas
Empregou-se o método de Soxhlet, segundo método IUPAC 1.122,26 com n-hexano (faixa de ebulição 68-70 ºC). Foi fixada a velocidade/quantidade de 6 a 8 sifonagens por hora à temperatura constante e com fluxo de gotejamento entre 150 e 200 gotas por segundo. Foram pesados 5 g da amostra em vidro de relógio. As extrações foram realizadas em dois períodos de tempo distintos (4 e 8 h). Após o término do período de extração e arrefecimento da miscela, foram retiradas alíquotas de 3 mL para análises de lipídios totais, acidez e índice de peróxido. O restante da miscela foi concentrado a vácuo em evaporador rotativo da marca Marconi (modelo TE - 120) a 40-45 ºC, sendo o solvente recuperado.
Além da metodologia de Soxhlet, foram empregados os métodos de Folch et al.,4 com clorofórmio e metanol na proporção de 2:1 (v/v); de Bligh e Dyer,3 com clorofórmio, metanol e água na proporção de 1:2:0,8 (v/v) e de Hara e Radin,20com n-hexano e isopropanol na proporção de 3:2 (v/v), em cujas miscelas, recolhidas sobre uma camada de sulfato de sódio anidro, foram determinadas a acidez e o índice de peróxido (AOCS 5a-40 e Cd 8b-80,27 respectivamente). A análise espectrofotométrica na faixa do ultravioleta foi realizada somente nas miscelas obtidas através da metodologia de Hara e Radin,20 modificada por Evangelista e Regitano-D'Arce,28 na qual a mistura n-hexano-isopropanol foi substituída pela de isoctano-isopropanol. Os métodos de Folch et al.4 e Bligh e Dyer3 sofreram algumas modificações relatadas a seguir.
O método de Folch et al.,4 utilizado neste trabalho, seguiu algumas modificações propostas por Christie.6 Foram pesados 5 g da amostra em erlenmeyer de 250 mL com tampa de vidro, adicionando-se inicialmente 25 mL de metanol. A homogeneização foi realizada por cerca de 5 min em mesa agitadora da marca Nova Técnica (modelo NT 145), com agitação vigorosa. Após esta etapa, foram adicionados 50 mL de clorofórmio e a agitação foi efetuada por mais 10 min (2 min para frango). A mistura foi filtrada a vácuo, sendo o resíduo sólido ressuspendido em 30 mL da solução de clorofórmio-metanol 2:1 (v/v) e homogeneizado por 5 min. Após filtração, o sólido foi lavado mais uma vez com 50 mL de clorofórmio e com 25 mL de metanol e a extração prosseguiu conforme a metodologia original.
Para a extração dos lipídios segundo o método de Bligh e Dyer,3 foram pesados aproximadamente 10 g de cada amostra. Em um erlenmeyer de 250 mL foram adicionados 50 mL de metanol, 25 mL de clorofórmio e 10 mL de água sobre a aveia (umidade de 11,88%). Para a carne de frango (umidade de 73,82%) foram seguidas as modificações propostas por Christie6 e Smedes e Thomasen29 que iniciou com homogeneização por 4 min em erlenmeyer de 250 mL dotado de tampa de vidro, com uma mistura de solvente consistindo de 10 mL de clorofórmio e 20 mL de metanol. Após completa homogeneização, uma única fase foi obtida e procedeu-se à adição de mais 10 mL de clorofórmio, seguida de agitação por 1 min. Filtrou-se a mistura a vácuo e o resíduo tissular foi re-homogeneizado com 10 mL de clorofórmio. Os filtrados foram combinados e seguiu-se o procedimento original. O erlenmeyer foi tampado e colocado em mesa agitadora da marca Nova Técnica (modelo NT 145) por 20 min. Em seguida, foram adicionados mais 25 mL de clorofórmio e 25 mL de solução de sulfato de sódio 1,5%. Agitou-se a mistura por mais 2 min. A solução com a amostra foi transferida para um funil de separação e o procedimento original foi obedecido.
Alíquotas das frações lipídicas obtidas das diferentes metodologias de extrações dissolvidas em éter de petróleo foram aplicadas em placas cromatográficas de silicagel 60 F 20 x 20 cm (Merck) com fase móvel éter de petróleo, éter etílico e ácido acético (80:20:2) conforme proposto por Nelson.12 A placa cromatográfica após eluição foi revelada com vapor de iodo e o Rf das manchas dos padrões e dos componentes das amostras foi determinado.
Todas as análises foram realizadas em triplicata. O delineamento experimental adotado foi o de blocos casualizados. Os dados obtidos foram submetidos à análise de variância e as médias foram comparadas entre si pelo teste de Tukey, (p < 0,05), utilizando-se o programa SAS.
RESULTADOS E DISCUSSÃO
Comparação dos métodos de extração
A Tabela 1 mostra os valores obtidos para os rendimentos em lipídios totais nas amostras e métodos testados. Apesar de o método de Bligh e Dyer3 ter apresentado o melhor rendimento dentre os métodos analisados, a metodologia de Soxhlet rendeu resultados que não ficaram aquém dos encontrados na literatura. É importante salientar que o solvente usado (n-hexano) é apolar e não tem a mesma eficiência para extrair os lipídios ligados (polares) como outros solventes de maior polaridade. No entanto, uma das vantagens deste método é a de que a amostra permanece boa parte do tempo imersa no solvente, ocorrendo sifonagens intermitentes que renovam o líquido constantemente, mantendo cada vez maior o gradiente de concentração de óleo entre solvente e amostra que possibilita a solubilização do óleo. A vantagem da imersão é a de que o uso do n-hexano como solvente tem sua eficácia aumentada, pois este solvente apolar não tem capacidade de extrair todos os lipídios mais polares, quando o processo é aplicado em um curto período de tempo. Na extração de Bligh e Dyer,3 a amostra também fica imersa em contato com os solventes por um período de tempo que não ultrapassa 15 min (agitação), dependendo do tipo de amostra, mas a mistura de solventes utilizada tem a capacidade de extrair um maior número de lipídios polares da amostra analisada.
O bom rendimento em lipídios totais, em ambas amostras analisadas (Tabela 1), proporcionado pelo método de Bligh e Dyer3 pode ser explicado pela ampla faixa de polaridade apresentada pela mistura de solventes utilizada. Clorofórmio e metanol são mais polares que n-hexano e isopropanol e, dessa forma, há uma extração eficiente de lipídios polares e apolares. Neste trabalho utilizou-se também a metodologia de Folch et al.4 que emprega a mistura clorofórmio e metanol, mas que não atingiu uma eficiência tão boa quanto a do método de Bligh e Dyer,3 provavelmente devido a perdas significativas nas etapas de lavagens sucessivas, recomendadas pelos autores, para a recuperação dos lipídios durante a extração. Outra desvantagem dessa metodologia é a desconsideração do conteúdo de água tissular no alimento analisado.
A extração de Bligh e Dyer3 pode sofrer algumas alterações, mas é imperativo que a proporção de clorofórmio, metanol e água seja mantida, antes (1:2:0,8) e após (2:2:1,8) a diluição com os solventes. Estas proporções representam os volumes totais existentes no sistema ternário formado pela mistura dos solventes, e inclui a água tissular do alimento estudado.
Uma característica interessante que o método de Bligh e Dyer3 apresentou durante o experimento foi produzir um rendimento superior aos demais métodos. Este poder de extração foi atribuído, entre outros fatores, ao processo de homogeneização vigorosa aplicado nesta metodologia. O poder de extração também tem íntima relação com o fato de que nesse método3 uma quantidade insuficiente de metanol e água esteve presente para a remoção dos não-lipídios, os quais são geralmente solubilizados pelos lipídios polares na fase orgânica.
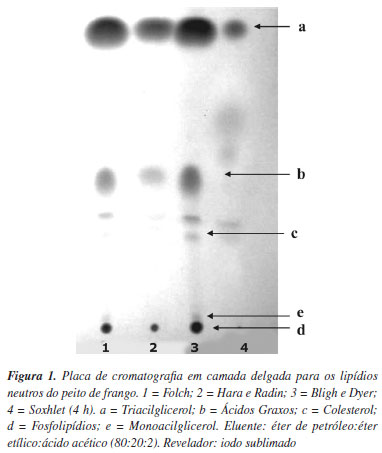
A identificação dos componentes lipídicos, nas placas cromatográficas, ocorreu por comparação dos valores de Rf dos padrões de monoacilgliceróis, triacilgliceróis, esteróis e ácidos graxos. Conforme as Figuras 1 e 2, o fosfolipídio, por ser um lipídio polar, não sofreu influência da fase móvel utilizada, portanto, a mancha que o identifica ficou retida no ponto de partida das amostras. Tanto na aveia em flocos quanto no peito de frango confirmou-se a presença dos lipídios neutros (monoacilgliceróis, triacilgliceróis, esteróis, ácidos graxos) em todas as metodologias de extração aplicadas, bem como a presença de fosfolipídios. Entretanto, o método de Hara e Radin apresentou menor intensidade (menor concentração) das manchas na placa cromatográfica. A presença de monoglicerídios não foi detectada com a utilização do sistema de solventes empregado. No caso dos fosfolipídios, menor intensidade das manchas foi visualizada tanto no método de Hara e Radin quanto no método de Soxhlet, em ambos os períodos de tempo, comprovando que o hexano, um solvente apolar, extrai com menor eficiência os lipídios polares, visto que a solubilidade dos lipídios polares é menor em solventes hidrocarbonetos, como o hexano, que em clorofórmio.6 A Tabela 2 apresenta os resultados de Rf's dos componentes lipídicos investigados.
Schäfer14 comparou as misturas de solventes isopropanol-hexano e clorofórmio-metanol na extração de lipídios de tecidos animais e vegetais. Este pesquisador verificou que a mistura clorofórmio-metanol obteve um melhor rendimento em lipídios totais quando a amostra em estudo foi o peito de frango, entretanto, quando a extração foi realizada em cereais, a mistura do álcool e um hidrocarboneto foi mais satisfatória que o outro método.
Youngs et al.30 investigaram a composição e distribuição dos lipídios em dois cultivares de aveia (Dal e Froker). Para obtenção das frações lipídicas foram utilizados os métodos de Soxhlet, com éter dietílico e os extratos foram extraídos novamente com n-butanol saturado em água. Através de cromatografia em camada delgada (CCD) a fração lipídica foi separada e caracterizada com obtenção de doze classes, sendo a maior porcentagem de triacilgliceróis (41%) seguida da presença de glicerídios, tais como monogalactosilglicerídios e digalactosilglicerídios (11%), e de fosfolipídios, como fosfatidilcolina, fosfatidiletanolamina e lisofosfatidiletanolamina (10% no total). Outros compostos encontrados em menor escala foram ácidos graxos livres (5%), diacilgliceróis (3%) e esteróis (1%).
Utilizando CCD para separar e quantificar doze componentes lipídicos de dois cultivares de aveia, Youngs et al.31 também confirmaram que a fração triglicerídica foi a mais abundante nas amostras analisadas, seguida da fração de digalactosilglicerídios. Uma pequena fração de esteróis foi identificada, porém não foi possível quantificá-la.
Undeland et al.16 confirmaram a eficiência do sistema clorofórmio-metanol-água na extração dos lipídios polares do arenque (Clu pea harengus), realizando análises de separação e quantificação das classes lipídicas. Esses lipídios polares, na sua maioria fosfolipídios, foram facilmente extraídos dos tecidos devido ao arranjo de bicamadas das membranas nas quais eles estão situados. Por conta desse arranjo, o qual expõe somente os lipídios polares, é compreensível o porquê da necessidade de solventes polares para extração.
De acordo com Bligh e Dyer3 e Christie,32 o sistema monofásico, obtido utilizando clorofórmio-metanol-água na proporção de 1:2:0,8 (v/v), possui as propriedades ideais para uma extração eficiente. Esse sistema supera as interações entre os lipídios e o tecido da matriz estudada e também os dissolve prontamente.
A baixa eficiência da mistura alcano-álcool-água para a extração de lipídios polares (fosfolipídios) pode ser explicada por duas razões: a primeira é que a parte polar dessa mistura (isopropanol) é menos polar que o metanol, o qual constitui a porção polar das metodologias propostas por Folch et al.4 e Bligh e Dyer.3 No entanto, o método de Hara e Radin,20 que utilizou a mistura n-hexano-isopropanol, apresentou vantagens como menor toxidez e melhor separação das fases (mais rápida) quando da adição do sulfato de sódio anidro, na etapa de lavagem, removendo eficientemente os não-lipídios da fase orgânica. A segunda razão é que há também o fato de que a solubilidade dos lipídios polares é menor em solventes hidrocarbonetos, tais como n-hexano, que em clorofórmio.16
No presente experimento, cada método apresentou vantagens e desvantagens. Com relação ao recolhimento da fase orgânica, o método de Hara e Radin20 foi mais conveniente que os métodos de Bligh e Dyer3 e Folch et al.,4 pois a fase orgânica foi separada na parte superior do sistema formado, em contraste com os sistemas que utilizam o clorofórmio. Esta vantagem está atrelada ao fato de não haver contaminação dos não-lipídios que estão na fase aquosa do sistema. A respeito da evaporação dos solventes, menos de 5 min foram necessários para a evaporação do clorofórmio, ao passo que mais de 10 min foram necessários para evaporar o n-hexano, quando se desejou obter os lipídios totais concentrados.
Qualidade da fração lipídica
Previamente à extração, as amostras de aveia em flocos e peito de frango não sofreram nenhum tipo de tratamento de secagem, pois isso levaria à lesão dos tecidos e também desencadearia o processo de oxidação do óleo. Sendo assim, os alimentos foram analisados na matéria úmida evitando-se maiores danos durante a extração.
A análise da acidez dos lípides extraídos da aveia em flocos (Tabela 3) pelo método de Soxhlet apresentou diferença significativa (2,2 a 2,2% AGL) em relação aos outros métodos (0,9 a 0,9% AGL), evidenciando, dessa forma, que, embora essa metodologia de extração apresente bons rendimentos, ela não deve ser aplicada quando se deseja utilizar o óleo obtido em análises posteriores.
Conforme a Tabela 3, a acidez aferida na fração lipídica do peito de frango não registrou valores muito elevados, ou seja, os métodos não afetaram severamente o material extraído. Entretanto, ocorreram diferenças nos valores entre as frações lipídicas produzidas pelo método de Soxhlet (1879) (2,6% AGL) e as demais metodologias (1,0% AGL). Essa diferença pode ser explicada pelo favorecimento da hidrólise dos triacilgliceróis durante o aquecimento das miscelas por tempo prolongado (4 e 8 h).
A determinação do estado oxidativo como reflexo da qualidade dos lipídios na matriz original em frações recuperadas após extração de Soxhlet ou outro método semelhante deve ser evitada,2,5,33 pois configura uma fonte de erro. Peróxidos podem ser formados durante o procedimento, já que há aquecimento constante da miscela e a exposição ao oxigênio durante a extração.
No experimento realizado, somente as frações lipídicas obtidas através da extração de Soxhlet apresentaram valores de peróxido, de 3,7 meq O2/kg para aveia em flocos e de 4,3 meq O2/kg para peito de frango (Tabela 4). As frações lipídicas das demais metodologias registraram valores iguais a zero, mostrando que o armazenamento das amostras sob congelamento não afetou a estabilidade oxidativa do óleo e que os métodos de extração a frio não são danosos à qualidade da matéria graxa extraída.
Com base nos dados obtidos nos índices de peróxidos da Tabela 4, conclui-se que apesar do óleo da aveia em flocos ser rico em ácidos graxos insaturados, os valores de peróxido (3,7 meq O2/1000 g), utilizando o método de Soxhlet, foram inferiores aos valores obtidos na fração lipídica do peito de frango (4,3 meq O2/1000 g). Uma provável explicação é a presença de antioxidantes naturais (ácidos ferúlico e caféico, tocóis e esteróis, p-hidroxibenzóico, p-hidroxifenilacético e p-cumárico) no óleo da aveia,34 que, de certa forma, minimizariam a degradação da fração lipídica, já que estes compostos reagem com os radicais livres, evitando a formação de peróxidos.
Em trabalho realizado concomitantemente no Laboratório de Óleos e Gorduras do Departamento de Agroindústria, Nutrição e Alimentos da ESALQ, Silva et al.35 verificaram a influência de diferentes doses de irradiação na estabilidade oxidativa da aveia em flocos. Foram avaliadas aveias em flocos irradiadas com 3,0; 4,5 e 6,0 kGy, durante um período de 6 meses. Os resultados da absortividade nos comprimentos de onda de 232 e 270 nm do ultravioleta que refletem a ocorrência da oxidação, através da quantificação dos dienos e trienos conjugados das frações lipídicas dos flocos de aveia irradiados ou não, não apresentaram um aumento significativo entre as doses, isto é, não ocorreu aumento na absortividade com o aumento da irradiação. Os valores de absortividade em 232 nm variaram entre 0,0933 a 0,1050 e entre 0,0445 a 0,0509 em 270 nm.
Esses resultados serviram de parâmetro para o presente trabalho, sendo que dessa forma não foi necessária a repetição das determinações. Os valores encontrados por Silva et al.35 ratificaram a inexistência de peróxidos nas extrações realizadas à temperatura ambiente.
No que diz respeito ao peito de frango, a formação de peróxidos foi, indubitavelmente, causada pelo aquecimento no processo de extração e recuperação do solvente, bem como pela presença de oxigênio no armazenamento e na extração, pois as frações lipídicas produzidas pelos outros métodos não apresentaram peróxidos.
Some-se a isso a presença de fosfolipídios contendo ácidos graxos altamente insaturados em produtos cárneos, os quais estão susceptíveis à oxidação, pois estão localizados no interior e nas paredes celulares do tecido gorduroso. Por essa razão os fosfolipídios são os primeiros compostos expostos à oxidação quando as células são lesadas.36
Em outro trabalho realizado também concomitantemente no Laboratório de Óleos e Gorduras do Departamento de Agroindústria, Nutrição e Alimentos da ESALQ, Pino et al.37 avaliaram a estabilidade oxidativa das amostras de peito de frango congelado utilizando o teste do TBA, segundo Tarladgis et al..38
O teste do TBA (ácido 2-tiobarbitúrico) é um método utilizado freqüentemente para detecção da oxidação lipídica, pois revela o nível de aldeídos presentes no óleo. Estes são produtos de quebra dos peróxidos (ou hidroperóxidos) e o ácido 2-tiobarbitúrico reage especificamente com o malonaldeído, resultando em uma solução colorida que pode ser determinada espectrofotometricamente.33
Os valores de TBA do peito de frango, encontrados pelas autoras, variaram entre 0,2972 a 0,8875 mg/kg. A análise de determinação do valor de ácido 2-tiobarbitúrico (TBA) demonstrou que não houve alteração significativa de sabor e odor na amostra, confirmando a ausência de peróxidos nas miscelas de Folch et al.,4 Bligh e Dyer3 e Hara e Radin.20
Os valores apresentados na Tabela 3 indicam que o processo de oxidação ocorreu com acúmulo de peróxidos (4,3 meq O2/kg) na metodologia de Soxhlet e através da análise de TBA pode-se afirmar que eles não foram quebrados a compostos secundários da oxidação (aldeídos), já que o valor máximo obtido por Pino et al.37 não ultrapassou 0,8875 mg/kg.
CONCLUSÕES
A metodologia escolhida para a extração da fração lipídica pode afetar a quantidade extraída e a sua qualidade oxidativa. Neste estudo, os solventes utilizados nas extrações e a temperatura influenciaram significativamente a eficiência da extração, de acordo com as especificidades e interações nas matrizes analisadas.
O solvente para extração de óleo de um tecido animal ou vegetal deve ser escolhido conforme as características intrínsecas (químicas e físicas) da matriz.
O método de Soxhlet, por ser de execução mais simplificada, pode ser recomendado tanto para amostras de origem animal como vegetal, quando não houver emprego posterior do extrato.
Extratos obtidos com hexano/isopropanol apresentam um menor rendimento em lipídios totais e uma capacidade insuficiente de extrair os componentes lipídicos polares das amostras analisadas.
Com base nos resultados deste trabalho, quando as frações lipídicas forem extraídas para uso posterior, recomenda-se a metodologia de Bligh e Dyer, tanto para amostras de origem vegetal como animal.
Recebido em 13/2/08; aceito em 20/10/08; publicado na web em 26/2/09
- 1. Regitano-d'Arce, M. A. B.; Lima, U. A.; Ciência e Técnologia de Alimentos 1987, 7, 1.
- 2. Kates, M.; Techniques of lipidology: isolation, analysis and identifica tion of lipids, Elsevier Applied Science: London, 1972, cap. 2.
- 3. Bligh, E.G.; Dyer, W.J.; Can. J. Biochem. Physiol 1959, 37, 911.
- 4. Folch, J.; Lees, M.; Stanley, G. H. S.; J. Biol. Chem. 1957, 226, 497.
- 5. Shahidi, F.; Wanasundara, J. P. D. Em Food lipids: chemistry, nutrition and biotechnology; Akoh, C. C; Min, D. B., eds.; Marcel Dekker: New York, 1998, cap. 5.
- 6. Christie, W. W.; Lipid analysis, Pergamon Press: Oxford, 1982, cap. 2.
- 7. Smedes, F.; The Analyst 1999, 36, 1711.
- 8. Soxhlet, F.; Polytechnisches J 1879, 232, 461.
- 9. Schneider, F. H.; Fette Seifen Anstrichmittel 1980, 80, 16.
- 10. Luque De Castro, M. D.; García-Ayuso, L. E.; Anal. Chim. Acta 1998, 369, 1.
- 11. Hanson, S. W. F.; Olley, J.; Biochem. J 1963, 89, 101.
- 12. Nelson, J.G. Em Analyses of fats, oils and lipoproteins; Perkins, E. G., ed.; American Oil Chemists' Society: Champaign, 1991, cap. 2.
- 13. Hole, M.; Taylor, K. D. A.; Food Chem. 1996, 55, 215.
- 14. Schafer, K.; Anal. Chim 1998, 358, 69.
- 15. Smedes, F.; Askland, T. K.; Mar. Pollu. Bull 1999, 38, 193.
- 16. Undeland, I.; Härröd, M.; Lingnert, H.; Food Chem. 1998, 61, 355.
- 17. Iverson, S. J.; Lang, S. L. C.; Cooper, M. H.; Lipids 2001, 36, 1283.
- 18. Manirakiza, P.; Covaci, A.; Schepens, P.; Journal of Food Composition and Analysis 2001, 14, 93.
- 19. Fishwick, M. J.; Wright, A. J.; Phytochemistry 1977, 16, 1507.
- 20. Hara, A.; Radin, N. S.; Anal. Biochem 1978, 90, 420.
- 21. Sahasrabudhe, M. R.; J. Am. Oil Chem. Soc. 1982, 59, 354.
- 22. Sahasrabudhe, M. R.; Smallbone, B. W.; J. Am. Oil Chem. Soc 1983, 60, 801.
- 23. Gunnlaugsdoltir, H.; Ackman, R.G.; J. Sci. Food Agric 1993, 61, 235.
- 24. Gandhi, A. P.; Joshi, K. C.; Krishna, J.; Paribar, D. C.; Srivastav, P. R.; Kawalkar, J.; Jain, S. K.; Tripathi, R. N.; Int. J. Food Sci. Technol. 2003, 38, 369.
- 25. Association of Official Analytical Chemists (AOAC); Official methods of analysis, 13th ed., Arlington, 2000.
-
26International Union of Pure and Applied Chemistry (IUPAC); Standard methods for the analysis of oils, fats and derivatives, 6th ed., Pergamon Press: Oxford, 1979.
- 27. American Oil Chemists' Society (AOCS); Official methods and recom mended practices, 5th ed., AOCS: Champaign, 1997.
- 28. Evangelista, C. M.; Regitano-D`Arce, M. A. B.; Grasas y Aceites 1999, 50, 22.
- 29. Smedes, F.; Thomasen, T. K.; Mar. Pollut. Bull 1996, 32, 681.
- 30. Youngs, V. L.; Püskülcü, M.; Smith, R. R.; Cereal Chem 1977, 54, 803.
- 31. Youngs, V. L.; Cereal Chem 1978, 55, 591.
- 32. Christie, W. W.; Gas chromatography and lipids, The Oily Press: Ayr, 1989, cap. 2, p. 11-42.
- 33. Rossel, J. B. Em Rancidity in foods; Allen, J. C.; Hamilton, R. J., eds.; Black Academic and Professional: Glasgow, 1994. cap. 2, p. 22-51.
- 34. Peterson, D. M.; Emmons, C. L.; Hibbs, A. H.; Journal of Cereal Science 2000, 33, 97.
- 35. Silva, R. C.; Regitano-d'Arce, M. A. B.; Sarmento, S. B.; Resumos do 18ş Congresso Brasileiro de Ciência e Tecnologia de Alimentos, Porto Alegre, Brasil, 2002.
- 36. Ranken, M. D. Em ref. 33, cap.11, p. 191-202.
- 37. Pino, L. M.; Regitano-d'Arce, M. A. B.; Racanicci, A. M. C.; Resumos do 19ş Congresso Brasileiro de Ciência e Tecnologia de Alimentos, Recife, Brasil, 2004.
- 38. Tarladgis, B. G.; Watts, B. M.; Younathan, M. T.; J. Am. Oil Chem. Soc 1960, 37, 44.
- 39. Olivo, R.; Shimokomaki, M.; Carnes no caminho da pesquisa; Cocal do Sul: Imprint: Cocal do Sul, 2001.
Datas de Publicação
-
Publicação nesta coleção
09 Jun 2009 -
Data do Fascículo
2009
Histórico
-
Aceito
20 Out 2008 -
Recebido
13 Fev 2008