Resumen
PEGylation has become a widely applied technique to enhancing in vitro and in vivo stability of therapeutic proteins and to increasing materials biocompatibility. PEG branched structures have proven useful for protein and peptide modification. Furthermore, they may be better than linear structures for many purposes. This paper describes an improved procedure for obtaining 2-arms PEG based on L-lysine. The efficiency of the synthesis was not related to moisture of the raw materials. This procedure does not use hazardous reagents as previous protocols do. It implemented a purification process for obtaining the desired structure with high purity ( > 99%). Finally, the procedure described here allows the obtaining of others PEGylation reagents.
PEGylation; branched PEG; interferon
PEGylation; branched PEG; interferon
ARTIGO
Un método reproducible para obtener peg biramificado monofuncional de alta pureza
A reproducible method for obtaining a 2-arms monofunctional peg with high purity
José A. RamónI,* * e-mail: jose@biomat.uh.cu ; Carlos PenicheI; Fidel R. CastroII; Vivian M. SáezII; Rosa M. ColásII; Rolando PáezII
ICentro de Biomateriales, Universidad de La Habana, Ave. Universidad e/ Ronda y G, Ciudad de La Habana, CP 10400, Cuba
IICentro de Ingeniería Genética y Biotecnología, Apdo. Postal 6162, Ciudad de La Habana, 10600, Cuba
ABSTRACT
PEGylation has become a widely applied technique to enhancing in vitro and in vivo stability of therapeutic proteins and to increasing materials biocompatibility. PEG branched structures have proven useful for protein and peptide modification. Furthermore, they may be better than linear structures for many purposes. This paper describes an improved procedure for obtaining 2-arms PEG based on L-lysine. The efficiency of the synthesis was not related to moisture of the raw materials. This procedure does not use hazardous reagents as previous protocols do. It implemented a purification process for obtaining the desired structure with high purity ( > 99%). Finally, the procedure described here allows the obtaining of others PEGylation reagents.
Keywords: PEGylation; branched PEG; interferon
INTRODUCCIÓN
El polietilenglicol (PEG) es un homopolímero sintético obtenido a partir del óxido de etileno. La habilidad única del PEG de ser soluble tanto en agua como en solventes orgánicos permite que pueda ser funcionalizado fácilmente y que la conjugación química a biomoléculas pueda hacerse en condiciones suaves que no afecten a éstas.1 Estudios del PEG en solución acuosa han demostrado que este polímero une 2-3 moléculas de agua por unidad de óxido de etileno. Debido a su alta flexibilidad y a la unión de las moléculas de agua el PEG actúa como si fuera 5-10 veces más grande que una proteína soluble de masa molecular comparable. Se ha sugerido que estas propiedades son las responsables de que el PEG: precipite las proteínas; excluya a las proteínas y células de las superficies; reduzca su inmunogenicidad y antigenicidad y prevenga la degradación de las proteínas por enzimas y células de mamíferos.2 Debido a estas características, la conjugación a polietilenglicol, conocida como PEGilación, se usa ampliamente para: aumentar la estabilidad in vitro e in vivo de proteínas terapéuticas, disminuir la adsorción de proteínas y células en la superficie de biomateriales, aumentar el tiempo de vida en el organismo de liposomas, entre otras muchas aplicaciones.2
Los primeros trabajos de PEGilación se realizaron en la década de los ´70. La posibilidad teórica de usar la conjugación a PEG de las proteínas, para disminuir la inmunogenicidad de éstas, se le ocurrió a Frank F. Davis en 1967 mientras preparaba la solicitud de financiamiento para un proyecto sobre aplicación terapéutica de proteínas no humanas.3 No obstante, no es hasta inicios de los ´90 cuando se aprueba el uso en humanos de las primeras proteínas PEGiladas (Adenosin Deamidasa y Catalasa de Hígado Bovino).4 Los estudios realizados durante los ´80 y los ´90 demostraron que la mayor utilidad de la PEGilación es en el aumento del tiempo de vida media en sangre de las proteínas conjugadas y no en la disminución de la inmunogenicidad de éstas.
Existen diversos procedimientos para obtener reactivos para PEGilar con variadas estructuras y grupos reactivos. La llamada "primera generación" estuvo compuesta por reactivos de estructura lineal y funcionalizados por grupos muy reactivos, por tanto poco específicos, como: diclorotriazina, tresilato, succinimidil carbonato, benzotriazol carbonato, p-nitrofenil carbonato, triclorofenil carbonato, carbonilimidazol, succinimidil succinato y acetaldehído.1 La elevada reactividad y baja especificidad de estos reactivos producía usualmente una PEGilación extensiva de las proteínas y se afectaba significativamente la actividad biológica de las mismas. No obstante, a partir de un miembro de esta generación, el succinimidil carbonato de PEG (SC-mPEG), se obtuvo un conjugado con interferón alfa 2b (Pegintron® de Schering-Pough Inc.) de probada efectividad en el tratamiento de la Hepatitis C y ventas anuales superiores a los 1000 millones de USD.5
En los años ´90 surge la llamada "segunda generación" de reactivos para PEGilar. En este grupo están comprendidos PEG funcionalizados con grupos de reactividad más específica como: aldehídos, para el amino del N-terminal de las proteínas y maleímido, para los grupos tioles. Además, en esta generación se adicionan los PEG ramificados.1
Los PEG ramificados tienen particularidades muy interesantes que los hacen extremadamente útiles en la conjugación de proteínas. Por ejemplo, unidos a las proteínas "actúan" como si fueran mucho más grandes que un PEG lineal de igual masa molecular. Esta estructura tiene la ventaja de que se unen dos cadenas de PEG a un mismo sitio de la proteína y se reduce así la posibilidad de su inactivación biológica. Además, debido a un fenómeno conocido como "efecto sombrilla", los PEG de varias ramas son más efectivos en reducir la antigenicidad e inmunogenicidad, proteger las proteínas de la proteólisis y disminuir la heterogeneidad y la filtración por riñón del conjugado que los PEG lineales de masa molecular semejante.1
Debido a que el PEG es un polímero lineal, para formar la estructura ramificada se necesita una molécula núcleo que posea los grupos funcionales necesarios para unir a dos o más cadenas de PEG y para formar el enlace, posteriormente, con las proteínas. El primer reactivo ramificado fue obtenido por Inada y colaboradores a partir de la triazina como núcleo.6
Yamasaki y colaboradores fueron los primeros en obtener una estructura ramificada más útil basada en la L-lisina como molécula núcleo.7 Posteriormente, Monfardini y colaboradores obtuvieron un PEG ramificado (conocido como PEG2) altamente purificado por unión de dos cadenas lineales de benzotriazol carbonato de PEG (o su semejante: SC-PEG) a los grupos α- y ε-amino de la L-lisina. Esta estructura permite obtener PEG de alta masa molecular (hasta 60 kDa) y con sólo un grupo funcional (el grupo carboxílico de la L-lisina).8
El reactivo descrito por Monfardini y colaboradores (Figura 1) ha sido utilizado con éxito en la conjugación de interferones alfa.9,10 La simplicidad de su estructura lo hace una opción interesante para diversas aplicaciones, desde la conjugación de proteínas hasta la de superficies de biomateriales, para aumentar su compatibilidad con los organismos vivos. Desafortunadamente, el procedimiento de síntesis descrito por estos autores es engorroso, peligroso (por ejemplo: usa fosgeno) y necesita materias primas de una calidad extremadamente alta.8
El objetivo de este trabajo fue obtener un PEG ramificado de dos ramas y monofuncional (estructura general descrita en la Figura 1) de forma eficiente, reproducible y con alta pureza. Se partió del procedimiento descrito por Monfardini y colaboradores8 pero se hicieron estudios de las distintas etapas para lograr el objetivo trazado.
De forma general, el procedimiento de Monfardini y colaboradores tiene tres etapas generales: obtención de un PEG lineal funcionalizado para reaccionar con grupos aminos; obtención de L-lisina biPEGilada y derivatización del grupo carboxílico de la L-lisina biPEGilada como éster de N-hidroxisuccinimida. En este trabajo se estudiaron y modificaron las etapas 1 y 2
Como grupo funcional, del PEG lineal, para la modificación de grupos amino se escogió el succinimidil carbonato. El SC-mPEG pertenece a la primera generación de reactivos para PEGilar.1 El grupo succinimidil carbonato reacciona fácilmente con grupos nucleofílicos como los aminos primarios, pero también con otros como los aminos imidazólicos y los hidroxilos. La especificidad hacia uno u otro grupo depende del pH pues, al tener distinto pK, la proporción de cada especie protonada no es la misma a determinada concentración de iones hidronio. Por ejemplo, en el caso de las proteínas, este reactivo reacciona a pH altos fundamentalmente por el grupo ε-amino de las L-lisinas pero a pH 6,5 reacciona principalmente por los grupos amino imidazólicos de las histidinas, aunque también reacciona con los hidroxilos de serinas y treoninas.9
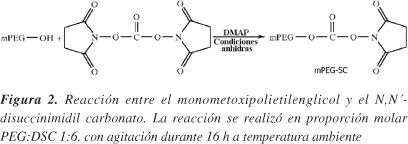
El procedimiento inicial de síntesis del SC-mPEG descrito era engorroso e incluso peligroso, pues el primer paso consistía en la obtención del cloruro de ácido de PEG por reacción del polímero con fosgeno.8 No obstante, Miron y Wilchek dieron a conocer un método muy sencillo para obtener este reactivo, basado en la reacción del mPEG-OH con DSC en presencia de DMAP (Figura 2) y posterior purificación del polímero funcionalizado por recristalización del precipitado de la mezcla reactiva por disolución en acetona y precipitación con éter dietílico.11
En este trabajo se estudiaron diversos factores que pueden influir en el rendimiento de la reacción anterior. Se buscaron condiciones de funcionalización que permitían una activación alta del mPEG-OH de forma reproducible; sobre todo en presencia de una alta humedad ambiental, fenómeno común en los países tropicales. Este aspecto es muy importante porque el SC-PEG pierde fácilmente el grupo funcional por hidrólisis11 por lo que todos los solventes utilizados en su síntesis deben estar bien secos y la técnicas operatorias utilizadas deben garantizar las condiciones anhidras.
PARTE EXPERIMENTAL
Materiales
Los monometoxipolietilenglicoles (mPEG-OH) fueron adquiridos en Nektar Therapeutics (Huntsville, AL, USA). El N,N´-disuccinimidil carbonato (DSC) y la 4-(dimetilamino)-piridina (DMAP) fueron suministrados por Sigma-Aldrich (Allentown, PA, USA). El interferón-α2b fue producido en el Centro de Ingeniería Genética y Biotecnología (Habana, Cuba). El resto de los reactivos y solventes fueron obtenidos de Merck (Darmstadt, Alemania).
Obtención de SC-PEG
Solubilidad del DSC
Cien mg de DSC se pesaron y se le añadió un mililitro del solvente correspondiente. La suspensión fue agitada fuertemente (Vortex, IKA, Alemania) durante 5 min. Posteriormente la muestra fue calentada en un baño termostatado (Buchi Laboretechnik AG, Flawil, Suiza) a 55 °C durante 10 min. Pasado este tiempo la muestra volvió a agitarse durante 5 min. La solubilidad se evaluó como desaparición total del sólido.
Secado de solventes
El 1,4-dioxano fue secado por agitación durante 16 h con sodio metálico y posterior destilación. El resto de los solventes, fue secado pasivamente sobre tamiz molecular (activado durante 12 h a 350 °C) durante 48 h.
Reacción de activación
Diez gramos de mPEG-OH se disolvieron en 250 mL de tolueno y se secaron por destilación azeotrópica durante 3 h. Pasado este tiempo el volumen se llevó a 100 mL y se dejó enfriar hasta temperatura ambiente. A la solución anterior se le añadió el cosolvente bien seco. Éste fue escogido entre: diclorometano (DCM, 60 mL), 1,4-dioxano (100 mL) y N,N-dimetilformamida (DMF, 100 mL). Posteriormente se añadieron 2 g de DSC disuelto en 15 mL de DMF y, gota a gota, 1 g de DMAP disuelto en 10 mL de DCM. Se dejó reaccionar durante 16 h con agitación. La mezcla de reacción se precipitó con 1 L de éter dietílico frío y el precipitado fue colectado por filtración al vacío o por centrifugación 30 min a 3000 rpm en una centrífuga Hitachi, Modelo CT6D (Hitachi, Tokio, Japón). Éste se recristalizó 3 veces por disolución en acetona a 35 °C y precipitación con éter dietílico. El producto final se secó con alto vacío y se almacenó bajo nitrógeno a -20 °C.
Reacción del SC-PEG y la L-lisina
Reacción en un paso
Doce gramos de SC-PEG se hicieron reaccionar con una solución de L-(+)-lisina (relación molar lisina:PEG 1:3) a 0,2 mg/mL en solución tampón borato 100 mmol/L, pH 8,5. La reacción se realizó a temperatura ambiente con agitación durante 16 h. Pasado este tiempo, la mezcla de reacción se diluyó 5 veces con agua bidestilada y se ajustó el pH a 3 con ácido clorhídrico. El PEG se extrajo 3 veces con igual volumen de DCM. La unión de las tres fracciones de extracción se secó con sulfato de sodio anhidro y se filtró. La solución de PEG en DCM se concentró hasta 20 mL en un rotoevaporador (Büchi Laboretechnik AG, Flawil, Switzerland). El concentrado se precipitó con 120 mL de éter dietílico frío, se separó por filtración y se secó a alto vacío.
Reacción en dos pasos
Seis gramos de SC-PEG se hicieron reaccionar con una solución de L-(+)-lisina (relación molar lisina:PEG 50:1) a 15 mg/mL en solución tampón borato 100 mmol/L, pH 8,5. La reacción se realizó a temperatura ambiente con agitación durante 4 h. Pasado este tiempo, la mezcla de reacción se diluyó 5 veces con agua bidestilada y se ajustó el pH a 3 con ácido clorhídrico. El PEG se extrajo 3 veces con igual volumen de DCM. La unión de las tres fracciones de extracción se secó con sulfato de sodio anhidro y se filtró. La solución de PEG en DCM se concentró hasta 20 mL en un rotoevaporador (Büchi Laboretechnik AG, Flawil, Switzerland). El concentrado se precipitó con 120 mL de éter dietílico frío, se separó por filtración y se secó a alto vacío. Este sólido se disolvió en DCM seco a 0,1 g/mL, se le añadieron 6 g de SC-PEG de igual masa molecular y el pH se ajusto a 8,5 con trietilamina. La reacción se realizó a temperatura ambiente con agitación durante 16 h. Luego la solución de PEG en DCM se concentró hasta 20 mL. El concentrado se precipitó con 120 mL de éter dietílico frío, se separó por filtración y se secó a alto vacío.
Procedimiento de purificación de la L-lisina biPEGilada (Lis-2PEG)
Una columna que contenía 1 L de la matriz cromatográfica DEAE-sefarosa se equilibró con 3 volúmenes de solución tampón borato 100 mmol/L, pH 7,5 y posterior lavado con 5 volúmenes de agua bidestilada. Diez gramos de la mezcla de reacción disueltos en agua bidestilada a 5 mg/mL se aplicaron a la columna. El PEG sin reaccionar se eliminó lavando la columna con 2 volúmenes de agua bidestilada y la Lis-2PEG se eluyó con una solución de cloruro de sodio 1 ó 10 mmol/L. A esta fracción se le ajustó el pH a 3 con ácido clorhídrico y se extrajo 3 veces con igual volumen de DCM. La unión de las tres fracciones de extracción se secó con sulfato de sodio anhidro y se filtró. La solución de PEG en DCM se concentró hasta 10 mL. El concentrado se precipitó con 60 mL de éter dietílico frío, se separó por filtración y se secó a alto vacío.
Estudio de elusión de Lis-2PEG
La muestra obtenida según el procedimiento de Monfardini y colaboradores,8 eluida con una solución 10 mmol/L de NaCl, se aplicó a una columna equilibrada como se describe en el epígrafe anterior. De forma sucesiva se aplicaron iguales volúmenes de agua y de solución de NaCl a: 100, 200 y 500 μmol/L; 1, 2, 5 y 10 mmol/L. La fracciones eluídas fueron analizadas por SDS - PAGE.
Activación de Lis-2PEG y conjugación a IFN- α2b
Activación del PEG
Seis gramos de Lis-2PEG se disolvieron en 20 mL de DCM seco y se le añadieron 60 mg de N-hidroxisuccinimida y 250 mg de N,N-diciclohexilcarbodiimida. La reacción se mantuvo bajo agitación durante 24 h a temperatura ambiente. Se filtró, se concentró hasta 5 mL y se precipitó con 20 mL de éter dietílico frío. El precipitado se recristalizó 3 veces disolviendo en acetona y precipitando con éter dietílico. El producto final se secó a alto vacío y se almacenó bajo nitrógeno a -20 °C.
Conjugación a IFN-α2b
Cien miligramos del PEG activado como éster de N-hidroxisuccinimida fueron añadidos a una solución que contenía IFN-±2b a 6 mg/mL (relación molar proteína:PEG, 1:3) en solución tampón borato 120 mmol/L, pH 8,5. La reacción se mantuvo durante 1 h a 4 °C con agitación suave. La reacción se detuvo diluyendo 50 veces con solución tampón acetato de sodio 10 mmol/L, pH 4.
Fraccionamiento de la reacción de conjugación
Una columna XK 26/40 (Pharmacia) que contenía 50 mL de Fractogel EMD 650 (M) COO- se equilibró a 10 mL/min con 150 mL de solución tampón acetato de sodio 10 mmol/L, pH 4. La solución que contenía la mezcla de reacción fue aplicada en la columna al mismo flujo. El PEG sin reaccionar fue lavado con 100 mL de la solución de equilibro y los conjugados con más de un residuo de PEG fueron eliminados con un lavado de 2 h con solución tampón acetato de sodio 50 mmol/L, pH 4 con 25 mmol/L de cloruro de sodio. El conjugado monoPEGilado fue eluído con solución tampón acetato de sodio 50 mmol/L, pH 4 con 150 mmol/L de cloruro de sodio.
Técnicas analíticas
Electroforesis en geles de poliacrilamida y en presencia de dodecil sulfato de sodio (SDS - PAGE)
Se utilizaron geles de gradiente (4-17% acrilamida) o lineales (8 ó 15% acrilamida).12 Los geles fueron teñidos específicamente para proteínas con Coomassie brilliant blue (CBB) y para PEG con yodo.13 La imágenes digitales de los geles fueron obenidas con un escaner ScanJet 4c/T (Hewlett Packard, Palo Alto, CA, USA) y analizadas con el software Molecular Analyst (Bio-Rad Laboratory, Hercules, CA, USA).
Determinación del grado de activación
La proporción de PEG activado se determinó por reacción con glicil-glicina y posterior cuantificación de los grupos amino por el método del ácido trinitrobenceno sulfónico (TNBS). De forma breve: un equivalente del PEG activado se hizo reaccionar con un equivalente de glicil-glicina en tampón borato 0,1 mol/L, pH 8, durante 1 h. Pasado este tiempo, se hizo reaccionar con TNBS en tampón borato 0,2 mol/L, pH 9,3, durante 30 min y se determinó la absorbancia de la muestra a 420 nm. El grado de activación se determinó por interpolación con una curva patrón.
RESULTADOS Y DISCUSIÓN
Obtención de SC-mPEG
Primero se estudió el solvente para disolver el DSC. En el procedimiento descrito por Miron y Wilchek, el DSC era adicionado en acetona11 donde sólo se logra obtener una suspensión. El hecho de que el DSC sea agregado totalmente disuelto a la mezcla reactiva es muy importante porque se evita una reacción en dos fases, que es menos eficiente que en fase homogénea. Además, la acetona tiene gran afinidad por el agua y en este caso la humedad asociada a este solvente era particularmente indeseable, porque iba a estar presente durante todo el proceso de activación y el agua hidroliza rápidamente al SC-mPEG. De los solventes estudiados (tolueno, DCM, acetona, acetonitrilo, DMF, tetrahidrofurano y 1,4-dioxano), el escogido fue el DMF, donde el DSC era soluble después de un suave calentamiento a 55 °C.
Se estudió entonces el efecto del solvente del DSC sobre el grado de activación (fracción porcentual de moléculas de mPEG-OH funcionalizadas como succinimidil carbonato) del SC-mPEG. El grado de activación fue significativamente superior cuando el DSC se disolvió en DMF que cuando se resuspendió en acetona (89±3% versus 73±3%, p<0,01). Suponemos que esta diferencia se debe esencialmente a la heterogeneidad de la reacción en el segundo caso, aunque también pueden incidir la dificultad en eliminar las trazas de humedad de la acetona.
El otro aspecto estudiado fue el de los solventes utilizados para preparar la mezcla de reacción. Miron y Wilchek utilizaron el 1,4-dioxano (secado sobre tamiz molecular) para disolver el mPEG-OH y acetona (también secada sobre tamiz molecular) para "disolver" el DSC y la 4-(dimetilamino)-piridina (DMAP).11 En este trabajo se introdujo un secado azeotrópico inicial del mPEG-OH con tolueno durante 3 h. Este paso es muy importante pues la gran afinidad por el agua del PEG provoca una rápida absorción de la humedad ambiental por este polímero. La derivatización del mPEG-OH como succinimidil carbonato sin un secado inicial del mPEG-OH es extremadamente ineficiente (resultados no mostrados). Por tanto, un solvente constante en nuestra mezcla reactiva fue el tolueno. El PEG es relativamente poco soluble en tolueno a temperatura ambiente por lo que se decidió utilizar un co-solvente que se añadió después del secado azeotrópico. Este fue escogido entre los siguientes: 1,4-dioxano, utilizado en el procedimiento inicial; DMF, solvente del DSC; y diclorometano, sustancia donde el PEG es muy soluble. La proporción de co-solvente utilizada fue la mínima necesaria para mantener al PEG disuelto a temperatura ambiente. La DMAP se añadió disuelta en una mezcla de solventes en iguales proporciones que en la mezcla reactiva. Los resultados (Tabla 1) mostraron que los grados de activación mejores se obtuvieron con el DCM y el 1,4-dioxano. No obstante, en lo sucesivo se trabajó con DCM para evitar el secado con sodio del 1,4-dioxano.
Finalmente se estudió el proceso de recristalización del PEG activado. Específicamente, el procedimiento de colección del polímero precipitado con éter dietílico. En el procedimiento descrito por Miron y Wilchek el precipitado era colectado por filtración.11 No obstante, la temperatura de ebullición del éter dietílico está muy cercana a la temperatura ambiente por lo que en un sistema abierto se evapora rápidamente y provoca una disminución de la temperatura del sistema y, por ende, la condensación de la humedad ambiental. Este fenómeno es perjudicial para la obtención de SC-mPEG debido a que el agua lo hidroliza fácilmente. En nuestro trabajo este problema adquirió una gran importancia cuando se realizaba la síntesis de cantidades de mPEG-OH superiores a los 20 g donde los pasos de filtración requirieron un tiempo largo. Para tratar de evitar esta dificultad se estudió el uso de la centrifugación para colectar el polímero precipitado. Los resultados de este estudio demostraron que al usar la centrifugación los grados de activación obtenidos eran inferiores que separando el precipitado por filtración (88±3% versus 53±5%, p<0,001). Una posible explicación para este resultado es que el agua presente como trazas en la acetona y el éter dietílico y/o absorbida de la atmósfera se acumula junto con el polímero en el fondo del frasco de centrifugación debido a su mayor densidad y provoca la desactivación del SC-mPEG. Para eliminar el problema de la absorción de humedad durante la filtración se decidió realizar esta operación en atmósfera de argón seco.
En conclusión, los estudios realizados permitieron diseñar un procedimiento (Figura 3) para obtener SC-mPEG de forma reproducible con un alto de grado de activación. Los resultados con éste resultaron ser independientes de la humedad inicial del mPEG-OH y del ambiente.
Obtención de Lis-2PEG
La obtención de Lis-2PEG consta de dos partes: reacción del aminoácido con las cadenas lineales del polímero funcionalizado y purificación del derivado bisustituido
Reacción de la L-lisina con el SC-mPEG
Monfardini y colaboradores describen dos procedimientos para obtener el derivado bisustituido de la L-lisina.8 El primero consta de un solo paso en medio acuoso y lo utilizaron para PEGilar la L-lisina con dos cadenas iguales. El otro consta de dos pasos: uno inicial en medio acuoso, para obtener un derivado monosustituido, y una segunda etapa en medio orgánico. Este proceso en dos etapas es descrito para obtener el aminoácido sustituido con dos cadenas distintas.
En este trabajo el objetivo era obtener una L-lisina conjugada a dos cadenas semejantes. En el procedimiento descrito para sintetizar este tipo de estructura por Monfardini y colaboradores, la L-lisina se hace reaccionar en una proporción molar 1:3 con el SC-mPEG aún cuando la relación teórica es 1:2. Esta cantidad adicional es necesaria porque paralelamente a la conjugación del aminoácido ocurre la hidrólisis a mPEG-OH del polímero funcionalizado. Por tanto, un tercio del SC-mPEG se pierde por diseño en este procedimiento. Debido al alto valor del mPEG derivatizado esta pérdida constituye una desventaja.
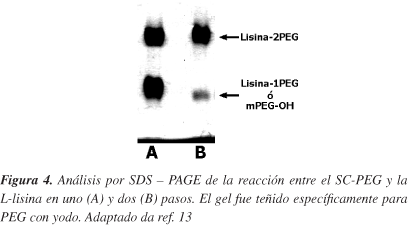
Se decidió entonces comparar los procedimientos en una y dos etapas, para la reacción entre la L-lisina y el SC-mPEG, respecto a la proporción porcentual del polímero en forma de aminoácido biPEGilado y recuperación de la masa inicial del PEG. A pesar que el protocolo en dos etapas tenía varios pasos más, era posible que el rendimiento global fuera mayor debido a que, por el diseño, el 100% del SC-mPEG se transforma en el aminoácido bisustituido (Parte Experimental). Los resultados de la comparación de ambos procedimientos (Figura 4) mostraron que el rendimiento con el procedimiento en dos etapas (Figura 7) es significativamente superior al de una, respecto a la cantidad de PEG en forma de L-lisina bisustituida (95±7% versus 56±6%, p<0,001).

Purificación de L-lisina biPEGilada
Según Roberts y colaboradores, la reacción de la L-lisina con los PEG lineales contiene las siguientes impurezas: mPEG-OH (procedente de la hidrólisis de los PEG lineales activados), L-lisina con una sola cadena de PEG (PEG1) y PEG3, el cual es el producto de un PEG biactivado, procedente de la fracción diol siempre presente en los monometoxiPEG, que unió una L-lisina en cada extremo y a su vez otras dos cadenas de PEG se unieron a los otros grupos amino (esta especie tiene dos grupos carboxílicos libres).1 No obstante, estos autores obviaron dos posibles impurezas también procedentes de los PEG biactivados producto de la fracción diol: un PEG con una L-lisina en cada extremo (PEG1+) y el formado por la impureza anterior que a su vez unió otra cadena de PEG a uno de los dos grupos amino libres (PEG2+). Así la mezcla de reacción contiene: una especie neutra (mPEG-OH), un zwitterión (PEG1), un doble zwitterión (PEG1+), un diácido (PEG3), uno monoácido-zwitterión (PEG2+) y el monoácido que es la especie de interés. Este último puede ser obtenido por una cuidadosa purificación mediante cromatografía de intercambio iónico.
El procedimiento descrito por Monfardini y colaboradores consistía en unir la mezcla de reacción disuelta en agua a una matriz de DEAE-sefarosa, equilibrada a pH ácido y a muy baja fuerza iónica. Después de lavar la matriz con agua, la L-lisina PEGilada era eluída con una solución de cloruro de sodio 10 mmol/L.8 Este procedimiento es relativamente sencillo pero en este trabajo sólo funcionó completamente bien cuando los contaminantes PEG1+, PEG2+ y PEG3 no estuvieron presentes en la mezcla de reacción. Cuando eso sucedió, los mismos coeluyeron con la estructura de interés (Figura 5-I, D). A pesar de que estos contaminantes estuvieron en una proporción muy pequeña, prácticamente indetectable, afectaron de modo sensible el posterior uso del PEG ramificado en la conjugación de proteínas. Por ejemplo, se conjugó el PEG purificado según el protocolo descrito por Monfardini y colaboradores con interferón α-2b y se realizó la separación cromatográfica de la mezcla obtenida (Figura 5-II). Se pudieron detectar una serie de señales que no se correspondieron con las especies esperadas: polímero y proteína sin reaccionar y los conjugados de distinta estequiometría.10 Muchas de las especies inesperadas eluyeron junto con el conjugado monoPEGilado (Figura 5-II, D) que es el que interactúa más fuertemente con la matriz de intercambio catiónico utilizada debido a su mayor contenido proteico. Por esta razón se consideró que esos "artefactos" contienen más de una molécula de IFN-α2b por lo que provienen de PEG biactivados. Dado que el PEG sin reaccionar (fracción B, Figura 5-II) muestra una sola banda correspondiente a la masa molecular del PEG de dos ramas, suponemos que la especie biactivada proviene de la impureza que se nombró PEG2+.
Para que los contaminantes PEG1+, PEG2+ y PEG3 no estén presentes en la mezcla de reacción el mPEG-OH inicial utilizado en la obtención del SC-mPEG debe tener una fracción de diol prácticamente igual a cero. Usualmente esto solo es posible para polímeros de masa molecular menor que 5 kDa.14 Debido a que el interés de este trabajo es utilizar cadenas lineales de más de 10 kDa, se diseñó un experimento para buscar condiciones de elusión que permitieran obtener la estructura de interés con elevado grado de pureza. La fracción eluída a 10 mmol/L de NaCl fue desalada y unida a la matriz DEAE-sefarosa en iguales condiciones que en el experimento anterior. Posteriormente se fue eluyendo con concentraciones crecientes de NaCl pero siempre por debajo de 10 mmol/L (Figura 6-I). La estructura de interés eluyó a una concentración de 1 mmol/L de NaCl (Figura 6-I, E y F) y los contaminantes a concentraciones cinco veces superiores (Figura 6-I, H, I y J) y se garantizó así la pureza del primero. Se observó que los contaminantes eluyeron a fuerzas iónicas superiores lo cual corroboró la hipótesis de que el contaminante principal es el nombrado como PEG2+. La estructura así purificada (según procedimiento descrito en la Figura 8) fue activada como éster de N-hidroxisuccinimida y conjugada a interferón α-2b con muy buenos resultados (Figura 6-II, C). Este conjugado ha sido utilizado recientemente en un ensayo clínico Fase I con resultados satisfactorios.
Si bien el procedimiento de reacción y purificación obtenido en este trabajo (Figura 7 y 8) permitió obtener la estructura ramificada de interés con elevado grado de pureza y de forma reproducible, su éxito depende de la masa molecular y de la procedencia del mPEG-OH usado como materia prima. Consecuentemente, resulta recomendable repetir el experimento descrito en el párrafo anterior para encontrar la condición óptima de elusión cada vez que se desee obtener una nueva estructura.
Funcionalización de la L-lisina
El método descrito por Monfardini y colaboradores para transformar el grupo carboxílico de la L-lisina biPEGilada en éster de N-hidroxisuccinimida es muy sencillo y eficiente.8 En las condiciones de este trabajo se lograron grados de activación superiores al 95% y una recuperación de la masa inicial de polímero superior al 90%.
CONCLUSIONES
En este trabajo se obtuvo de forma eficiente, reproducible y con alta pureza la estructura de polietilenglicol descrita en la Figura 1. Se obtuvieron procedimientos optimizados para la síntesis de dos reactivos intermediarios (Figura 2, 7 y 8), cuyas eficiencias resultaron independientes de las condiciones ambientales.
Los resultados aquí mostrados no sólo son aplicables a la obtención de la estructura de la Figura 1. Por ejemplo: el SC-mPEG es un reactivo importante para la PEGilación de proteínas y biomateriales en general1,9,14 y los estudios de purificación son aplicables a cualquier estructura ramificada de PEG. Además, este compuesto ha sido utilizado como precursor de estructuras de mayor grado de ramificación como un PEG semejante a dendrímero desarrollado recientemente en nuestro grupo.10
Recebido em 28/5/08; aceito em 4/2/09; publicado na web em 3/7/09
- 1. Roberts, M. J.; Bentley, M. D.; Harris, J. M.; Adv. Drug Deliv. Rev. 2002, 54, 459.
- 2. Harris, J. M.; Chess, R. B.; Nat. Rev. Drug Discov. 2003, 2, 214.
- 3. Davis, F.; Adv. Drug Deliv. Rev. 2002, 54, 457.
- 4. Hinds, K. D.; Kim, S. W.; Adv. Drug Deliv. Rev. 2002, 54, 505.
- 5. Napoli, N.; Giannelli, G.; Antonaci, A.; Antonaci, S.; J. Viral. Hepat. 2008, 15, 300.
- 6. Matsushima, A.; Nishimura, H.; Ashihara, Y.; Yakata, Y.; Inada, Y.; Chem. Lett. 1980, 773.
- 7. Yamasaki, N.; Matsuo, A.; Isobe, H.; Agric. Biol. Chem. 1988, 52, 2125.
- 8. Monfardini, C.; Schiavon, O.; Caliceti, P.; Morpurgo, M.; Harris, J. M.;Veronese, F. M.; Bioconjugate Chem. 1995, 6, 62.
- 9. Wang, Y. S.; Youngster, S.; Grace, M.; Bausch, J.; Bordens, R.; Wyss, D. F.; Adv. Drug Deliv. Rev. 2002, 54, 547.
- 10. Ramon, J.; Saez, V.; Baez, R.; Aldana, R.; Hardy, E.; Pharm. Res. 2005, 22, 1374.
- 11. Miron, T.; Wilchek, M.; Bioconjugate Chem. 1993, 4, 568
- 12. Laemmli, U. K.; Nature 1970, 227, 680.
- 13. Kürfurst, M. M.; Anal. Biochem. 1992, 200, 244.
- 14. Veronese, F. M.; Biomaterials 2001, 22, 405.
Fechas de Publicación
-
Publicación en esta colección
22 Set 2009 -
Fecha del número
2009
Histórico
-
Recibido
28 Mayo 2008 -
Acepto
04 Feb 2009