Resumo
Monoacilglycerides and diacilglycerides are produced through lipase-catalyzed glycerolysis of soybean oil using Candida antarctica B in a solvent-free system. The reaction was carried out at a glycerol to triacylglycerol molar ratio of 8:1 with 2% of lipase. Acylglycerides, free fatty acids (FFA) and glycerol produced were separated employing the molecular distillation process. Starting from a product of enzymatic reaction 25.06% of triacylglycerols, 46.63% of diacylglycerides, 21.72% of monoacylglycerides, 5.38% of FFA and 1.21% of glycerol and after consecutively distillations, monoacylglycerides with 80% of purity was obtained and also oil with 54% of diacylglycerides to be used in human dietary.
monoacylglycerol; diacylglycerol; molecular distillation
monoacylglycerol; diacylglycerol; molecular distillation
ARTIGO
Produção de monoacilgliceróis e diacilgliceróis via glicerólise enzimática e destilação molecular
Production of monoglycerides and diglycerides through lipase-catalyzed glycerolysis and molecular distillation
Patricia Bogalhos Lucente Fregolente* * e-mail: lucente@feq.unicamp.br ; Gláucia Maria F. Pinto; Maria Regina Wolf-Maciel; Rubens Maciel Filho; César Benedito Batistella
Faculdade de Engenharia Química, Universidade Estadual de Campinas, 13081-970 Campinas - SP, Brasil
ABSTRACT
Monoacilglycerides and diacilglycerides are produced through lipase-catalyzed glycerolysis of soybean oil using Candida antarctica B in a solvent-free system. The reaction was carried out at a glycerol to triacylglycerol molar ratio of 8:1 with 2% of lipase. Acylglycerides, free fatty acids (FFA) and glycerol produced were separated employing the molecular distillation process. Starting from a product of enzymatic reaction 25.06% of triacylglycerols, 46.63% of diacylglycerides, 21.72% of monoacylglycerides, 5.38% of FFA and 1.21% of glycerol and after consecutively distillations, monoacylglycerides with 80% of purity was obtained and also oil with 54% of diacylglycerides to be used in human dietary.
Keywords: monoacylglycerol; diacylglycerol; molecular distillation.
INTRODUÇÃO
Atualmente, a conversão de gorduras e óleos em produtos de alto valor agregado, como monoacilgliceróis, ácidos graxos livres e derivados tem sido de grande interesse comercial. Os monoacilgliceróis (MAG) equivalem a 70% dos emulsificantes utilizados nas indústrias alimentícias, cosméticas e farmacêuticas. Além dos MAG, os diacilgliceróis (DAG) apresentam aplicação como emulsificantes não-iônicos e agentes texturizadores.1 Além das características emulsificantes dos DAG similares aos MAG por serem moléculas anfifílicas, os DAG são estruturalmente similares aos triacilgliceróis (TAG), elementos majoritários ( >95 %) nos óleos vegetais. Assim sendo, o óleo essencialmente de DAG pode ser incorporado na dieta humana uma vez que estes também apresentam propriedades similares ao óleo de TAG em relação ao cozimento de alimentos e estabilidade a altas temperaturas sem degradação do óleo, no caso de frituras.2,3 Pesquisas realizadas com ratos relatam que, ao contrário dos TAG que em excesso no organismo são estocados em forma de gordura localizada, os DAG são facilmente utilizados pelo organismo como fonte de energia.4,5 No Japão, estudos clínicos têm demonstrado que o uso de óleo de DAG em substituição ao óleo de TAG não somente reduz a gordura acumulada na região abdominal como previne doenças arterioscleróticas, obesidade e resistência à insulina.6,7
Industrialmente, MAG e DAG são produzidos através da interesterificação de triacilgliceróis com glicerol (glicerólise), a altas temperaturas ( >200 °C), na presença de catalisadores inorgânicos. Esta reação produz uma mistura com, aproximadamente, 50% de MAG.8 Devido às altas temperaturas utilizadas, esta reação é acompanhada pela formação de subprodutos indesejáveis, decorrentes de reações de polimerização de ácidos graxos. Estes componentes conferem cor escura e odor desagradável ao óleo.9
Com o crescente interesse das indústrias alimentícias em fabricar produtos mais saudáveis devido às exigências de mercado, ou seja, produtos livres de reações de polimerização e de gorduras na configuração trans, a substituição da rota química pela rota enzimática na produção de MAG e DAG apresenta-se como uma alternativa atrativa. Neste processo são utilizadas condições amenas de temperatura (40-70 ºC), não havendo reações paralelas de polimerização e alterações trans nas moléculas produzidas. Além disso, no processo enzimático é possível explorar a seletividade das enzimas em relação aos ácidos graxos e sua regiosseletividade pela posição do ácido graxo na molécula de glicerol (posição α ou β).10
Para separação e purificação dos acilgliceróis (TAG, DAG, MAG, AGL e glicerol) formados na reação enzimática, a técnica de destilação convencional não é recomendada, pois esses compostos podem sofrer degradação e oxidação quando expostos às elevadas temperaturas. Neste caso, a DM é a técnica mais apropriada, pois consiste em um processo para separação e purificação de substâncias termicamente sensíveis, assim como de líquidos com baixa pressão de vapor.11 Este método de separação não se trata de uma destilação propriamente dita, pois nesta técnica a separação dos compostos ocorre pela diferença de suas massas moleculares, e não através de equilíbrio entre fases. Assim sendo, a DM é considerada um processo de não equilíbrio.12,13
Fregolente e colaboradores,14 empregando a técnica de DM, obtiveram uma concentração de MAG de 96,3% efetuando várias destilações consecutivas a partir de uma mistura de acilgliceróis e ácidos graxos livres contendo inicialmente 43,6% de MAG na composição.
A DM tem sido aplicada com sucesso na área de lipídeos; além da concentração de MAG, um vasto número de trabalhos tem sido publicado na área, como recuperação de carotenóides a partir do óleo de palma,15 recuperação de tocoferol a partir do destilado da desodorização do óleo de soja,16 purificação e desodorização de lipídeos estruturados,17 preparação de concentrados de ácidos graxos poliinsaturados, 18 além de outros.
Este trabalho teve como objetivo separar os produtos obtidos na glicerólise enzimática do óleo de soja utilizando a técnica de destilação molecular visando obter dois tipos de materiais igualmente importantes: MAG destilados com propriedades emulsificantes e óleo enriquecido de DAG.
PARTE EXPERIMENTAL
Materiais
Neste trabalho foi utilizado óleo de soja refinado comercial, apresentando majoritariamente os seguintes ácidos graxos: 11,6% de C16:0, 22,5% de C18:1 e 54,9% de C18:2, glicerina (99%) (marca Synth), padrões para cromatografia gasosa, adquiridos da Sigma-Aldrich e lipase de Candida antactica B (Novozym 435) imobilizada cedida gentilmente pela Novozymes A/S (Bagsvaerd, Denmark).
Reação de glicerólise do óleo de soja catalisada por lipases
As reações de glicerólise enzimática livre de solventes foram realizadas num sistema em batelada em reator de vidro encamisado com saídas para retirada de amostras e medição de temperatura, com capacidade máxima de 1,0 L de reação. A mistura de substrato consistia de uma mistura isenta de solventes contendo glicerina (GL) e óleo de soja numa razão molar GL/TAG igual a 8, concentração de água de 3,5% em relação à massa de glicerol e quantidade de lipase de Candida antarctica B imobilizada de 2% em relação à massa de óleo. Assim sendo, para 1,0 L de reação as massas de reagentes foram: 500 g de óleo de soja, 432 g de glicerol, 10 g de lipase e 15,12 g de água. A temperatura foi mantida a 70 ºC, sob agitação de 300 rpm. As reações foram realizadas em triplicata por um período máximo de 24 h.19
Ao final desse período o meio reacional foi transferido para um funil de separação e deixado em repouso para a separação das fases. A fase aquosa (contendo também glicerol e as enzimas) foi separada da fase orgânica contendo os componentes MAG, DAG, AGL e TAG.
Análise de acilgliceróis e ácidos graxos
A análise de acilgliceróis e AGL foi efetuada por meio de cromatografia por exclusão de tamanhos (high-performance size exclusion chromatography, HPSEC), baseando-se no trabalho de Schoenfleder.20 O sistema cromatográfico utilizado consiste de uma bomba isocrática, modelo 515 HPLC pump (Warters), um detector de índice de refração modelo 2410 (Waters) e um módulo de controle de temperatura (Waters). As amostras foram injetadas utilizando-se um injetor manual, modelo Rheodyne 7725i com um loop de 20 μL (Waters). Duas colunas, Styragel HR 0.5 e HR 2 (Waters) de 7,8 x 300 mm de dimensão e tamanho de partícula de 5 μm foram posicionadas em série. A primeira coluna possui uma capacidade de separação de moléculas de massa molar na faixa de 100 até 5000 e a segunda, de 500 até 20000.
A fase móvel utilizada foi tetra-hidrofurano grau HPLC (Tedia) a uma vazão de 1 mL/min. Para esta vazão, a pressão típica de operação foi de 450 psi (3102 kPa). Todas as análises foram efetuadas a 40 ºC.
Destilação molecular
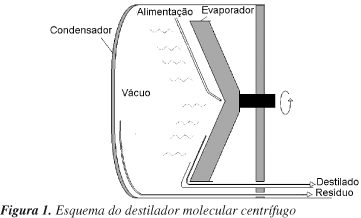
Foi utilizado um destilador molecular centrífugo da marca Myers Vaccum Inc. com área de evaporação igual a 0.0046 m2. Um esquema simplificado deste equipamento é mostrado na Figura 1. A mistura a ser destilada foi aquecida até a temperatura de alimentação (TA) de 60 ºC e através de uma bomba dosadora foi alimentado no centro do evaporador a uma vazão pré-determinada. Através da força centrífuga, um fino filme líquido forma-se uniformemente na superfície do evaporador, o qual foi aquecido por resistências elétricas. A velocidade do evaporador foi de 1350 rpm/min. A temperatura do evaporador (TE) variou de 100 a 250 ºC. Uma fração mais leve do material a ser destilado foi evaporada na superfície do evaporador, a qual atingiu a superfície do condensador aquecida. A temperatura do condensador (TC) foi mantida em 60 ºC, assim como as temperaturas das correntes de saída de destilado e resíduo (TD e TR, respectivamente). A Pressão (P) da câmara de vácuo atingiu 0,10 mmHg.
RESULTADOS
Glicerólise enzimática
Na etapa de glicerólise enzimática foram produzidos os acilgliceróis de diferentes massas moleculares a partir do óleo de soja para posterior separação e purificação dos compostos MAG e DAG. A lipase de Candida antarctica B, escolhida para atuar como biocatalisador da glicerólise enzimática isenta de solventes orgânicos, foi estudada juntamente com outras lipases, em relação à quantidade de lipase na reação, quantidade de água inserida no meio reacional e razão molar entre glicerol e óleo.19 Foi verificado que dentre as lipases estudadas, a lipase de Candida antarctica B obteve melhor desempenho na produção de DGA e MAG. A Figura 2 ilustra a conversão em fração mássica dos acilgliceróis em função da concentração da lipase de Candida antarctica B ao final de 24 h de reação. As conversões obtidas para cada reação de glicerólise quando utilizados 2, 5 e 10% de lipase no meio reacional são, respectivamente, 74, 78 e 88%. A Figura 2 mostra que a utilização de uma concentração de 10% de lipase no meio reacional aumenta em 30% a produção de MAG. Em contrapartida, diminui em 23% a produção de DAG e aumenta drasticamente a produção de AGL, de 3,36% (quando utilizados 2% de lipase) para 16,76% de AGL (quando utilizados 10% de lipase no meio reacional). As vantagens de se obter maior conversão de reação (88%) e maior produção de MAG quando se utiliza 10% de Candida antarctica B não supera as desvantagens de se empregar altas quantidades de lipase na reação (visto que o preço das lipases é o fator dominante quando se trata de custo do processo enzimático) e da alta produção de AGL, pois a separação dos MAG e AGL é dificultada mesmo utilizando a técnica de destilação molecular. Sendo assim, a concentração de 2% de Candida antarctica B foi fixada para produzir MAG e DAG para as etapas de destilação molecular.
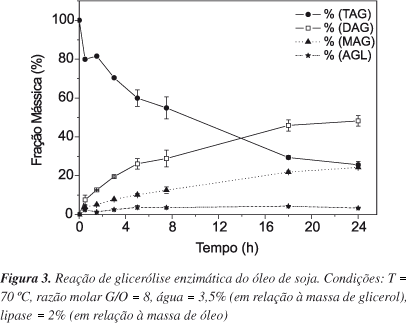
A Figura 3 mostra o comportamento cinético da reação de glicerólise enzimática do óleo de soja em função do tempo, para todos os componentes da mistura de acilgliceróis (TAG, DAG, MAG, AGL e GL) empregando 2% de lipase de Candida antarctica B. A mistura de acilgliceróis e AGL de diferentes massas molares obtida na reação de glicerólise enzimática em 24 h foi admitida como sendo a alimentação inicial das etapas de destilação molecular contendo 25,6% de TAG; 48,22% de DAG; 24,2% de MAG; 4,0% de AGL e 1,21% de GL.
Destilação molecular
A destilação molecular dos acilgliceróis foi realizada em sistema tipo "cascata", com realimentação do destilador com o material obtido na corrente de resíduo. Desse modo, a máxima recuperação de MAG na corrente de destilado pode ser alcançada, diminuindo-se gradativamente a concentração deste composto na corrente de resíduo.
As frações mássicas do material destilado (D) e do material recolhido na saída de resíduo (R) na pré-destilação são verificadas na Tabela 1. O material recolhido na linha de resíduo a 100 ºC foi novamente alimentado no destilador e, em seguida, a temperatura do evaporador (TE) foi elevada para 150 ºC.
Após a pré-destilação a 100 e 150 ºC para eliminar e/ou diminuir as concentrações de GL e AGL na corrente de resíduo, a TE foi aumentada de 190 até 250 ºC, gradativamente a cada etapa, juntamente com variações nas vazões de entrada (Q). A partir de TE =190º, foram realizadas destilações variando-se a vazão de entrada entre 6,0 e 10,0 mL/min, para verificar a influência do fluxo de alimentação no evaporador, além da influência da TE nas frações destiladas (Figura 4).

Verifica-se nas Figuras 4a e 4b que quanto maior a TE, porcentagens maiores de MAG e DAG são obtidas na corrente de destilado, em função da maior transferência de calor entre o evaporador e o fluido, permitindo que moléculas com massas molares mais elevadas sejam também evaporadas. Observou-se ainda que a transferência de calor foi mais efetiva quando a vazão de entrada (Q) era mais elevada, neste caso 10 mL/min. Isso porque uma menor vazão de entrada pode ocasionar formação de caminhos preferenciais na superfície do evaporador, ao invés de um filme homogêneo, negligenciando assim a transferência de calor da superfície do evaporador para o fluido. Com o aumento de Q para 10 mL/min, o fluido foi espalhado homogeneamente sobre a superfície do evaporador, proporcionando uma eficiente troca de calor do evaporador com o fluido a ser evaporado. Um aumento de Q de 6 para 10 mL/min, no caso de TE de 190 ºC, provocou um aumento de 31,88% de MAG na corrente de destilado.
Verifica-se na Figura 4a que, da mesma forma, os aumentos de Q nas temperaturas de 230 e 250 ºC provocaram aumentos de 16,8 e 4,0% de MAG destilados. No entanto, essa diferença na obtenção de MAG e DAG na corrente de destilado tanto a 6 como a 10mL/min foi reduzida com o aumento da TE, pois a temperatura do evaporador é alta o suficiente para que haja uma boa troca de calor, mesmo quando não há o completo recobrimento do fluido sobre o evaporador devido à formação dos caminhos preferenciais em Q menores.
Através da Figura 4a pode-se observar que a maior concentração de 80% de MAG destilados (DMG) foi obtida com uma vazão de alimentação de 10,0 mL/min e TE = 250 ºC, partindo-se de uma alimentação que continha, nesta etapa, inicialmente apenas 13,20% de MAG. A corrente de resíduo apresentou concentrações de MAG (RMG) não destilados nas condições experimentais.
A composição da corrente de alimentação foi continuamente alterada a cada aumento de TE. Sendo assim, a recuperação de MAG em cada etapa de destilação é diferente. O cálculo de recuperação de um determinado componente é determinado pela Equação 1:

sendo N o componente escolhido e R a porcentagem de recuperação de N.
As recuperações de MAG, bem como as concentrações de DMG, a diferentes temperaturas e vazões de alimentação são apresentadas na Tabela 2.
Nota-se que a recuperação de MAG é sempre maior quando Q é menor. Isto se deve ao tempo de residência das moléculas na superfície do evaporador: a concentração de DMG é diminuída quando Q é menor porque todas as moléculas apresentam disponibilidade de evaporação, diluindo a concentração (%) de MAG na corrente de destilado. Quando Q é aumentada, o tempo de cada componente na superfície do evaporador diminui e somente as moléculas mais propícias a evaporar nas condições fixadas como, por exemplo, os MAG evaporam, aumentando assim a concentração e a recuperação das moléculas mais leves na corrente de destilado.
Em contrapartida, a Figura 4b apresenta as concentrações de DAG destilados e residuais, nas condições estudadas. A quantidade de DAG destilada é baixa (mínimo de 1,0 e máximo de 8,1%), com um aumento em TE mais elevadas, de 230 a 250 ºC. Possivelmente, os DAG destilados eram constituídos de moléculas menores, com cadeias de ácidos graxos mais leves como, por exemplo, cadeias de ácido palmítico (C16).
O óleo resultante da corrente de resíduo após todas as etapas de destilação, conforme a Figura 5, apresentou composição majoritária de DAG da ordem de 54%. A composição do óleo de DAG apresentou 30,57% de TAG, 54,03% de DAG, 14,19% de MAG, 1,21% de AGL e 0% de GL. Além disso, o óleo de DAG obtido após todos os estágios de destilação apresentou coloração e odor semelhante ao óleo de soja original, mesmo após os aquecimentos a que foi submetido durante o processo. As propriedades do óleo de DAG foram preservadas, uma vez que a destilação molecular realizada em destilador centrífugo permite que o óleo a ser destilado permaneça em contato com o evaporador durante o tempo equivalente a 0,2-1 s.2 Este curto tempo de exposição do óleo às altas temperaturas faz com que as moléculas a serem destiladas sejam evaporadas sem ocasionar degradação excessiva do material, oxidação demasiada ou perda das características que tornam o óleo de DAG um potencial substituto do óleo de TAG na dieta humana.
CONCLUSÕES
Através de reações de glicerólise enzimática do óleo de soja foram obtidos os acilgliceróis MAG, DAG, TAG, AGL e GL que foram separados por destilação molecular a fim de concentrar e purificar os agentes emulsificantes MAG e obter um óleo concentrado em DAG utilizando a mesma técnica de separação. A lipase de Candida antarctica B imobilizada foi utilizada na obtenção dos acilgliceróis num sistema livre de solventes. Ao final de 24 h de reação enzimática, a mistura reacional apresentou 25,6% de TAG, 48,22% de DAG, 24,2% de MAG, 4,0% de AGL e 1,21% de GL. A mistura de acilgliceróis obtida após a reação de glicerólise foi então alimentada no destilador molecular para separação dos componentes e purificação dos MAG.
Recebido em 19/8/08; aceito em 20/1/09; pubicado na web em 3/7/09
- 1. Meng, X.; Zou, D.; Shi, Z.; Duan, Z.; Mao, Z.; Lipids 2006, 39, 37.
- 2. Hickman, K. C. D.; Chem. Rev. 1943, 34, 51.
- 3. Watanabe, H.; Onizawa, K.; Naito, S.; Taguchi, H.; Goto, N.; Nagao, T.; Matsuo, N.; Tokimitsu, I.; Yasukawa, T.; Tsushima, R.; Shimasaki, H.; Itakura, H.; Ann. Nutr. Metab. 2001, 45, 264.
- 4. Maki, C. K.; Davidson, M.; Tsushima, R.; Matsuo, N.; Tokimitsu, I.; Umporowicz, D. N.; Dicklin, M. R.; Foster, G. S.; Ingram, K. A.; Anderson, B. D.; Frost, S. D.; Bell M.; Am. J. Clin. Nutr. 2002, 76, 1230.
- 5. Yasunaga, K.; Glinsmann, W. H.; Seo, Y.; Katsuragi, Y.; Kobayashi, S.; Flickinger, B.; Kennepohl, E.; Yasukawa, T.; Borzelleca, J.; Food Chem. Toxicol. 2004, 42,1419.
- 6. Shimizu, M.; Moriwaki, J.; Nishide, T.; Nakajima, Y.; J. Am. Oil Chem. Soc. 2004, 81, 571.
- 7. Yamamoto, K.; Takeshita, M.; Tokimitsu, I.; Watanabe, H.; Mizuno, T.; Asakawa, H.; Tokunaga, K.; Tatsumi, T.; Okazaki, M.; Yagi, N.; Nutrition 2006, 22, 23.
- 8. Li, D.; Xu, T.; Takase, H.; Tokimitsu, I.; Zhang, P.; Wang, Q.; Yu, X.; Zhang, A.; Clinical Nutrition 2008, 27, 203.
- 9. Sonntag, N. O. V.; J. Am. Oil Chem. Soc. 1982, 59,795.
- 10. Ferreira-Dias, S.; Correia, A. C.; Baptista, F. O.; Fonseca, M. M. R.; J. Mol. Catal. B: Enzym. 2001, 11, 699.
- 11. Guo, Z.; Xu, X.; Green Chem. 2006, 8, 54.
- 12. Micov, M.; Lutisan, J.; Cvengros, J.; Sep. Sci. Technol. 1997, 32, 3051.
- 13. Batistella, C. B.; Maciel, M. R. W.; Comput. Chem. Eng. 1996, 20, 19.
- 14. Fregolente, L. V.; Batistella, C. B.; Maciel, R.; Wolf-Maciel, M. R.; J. Am. Oil Chem. Soc 2005, 82, 673.
- 15. Batistella, C. B.; Moraes, E. B.; Maciel Filho, R.; Maciel, M. R. W.; Appl. Biochem. Biotechnol. 2002, 98, 1149.
- 16. Moraes, E. B.; Batistella, C. B.; Alvarez, M. E. T.; Maciel Filho, R.; Maciel, M. R. W.; Appl. Biochem. Biotechnol. 2004, 113, 689.
- 17. Breivik, H.; Haraldsson, G. G.; Kristinsson, B.; J. Am. Oil Chem. Soc 1997, 74, 1425.
- 18. Kwon, S. J.; Han, J. J.; Rhee, J. S.; Enzyme Microb. Technol. 1995, 17, 700.
- 19. Fregolente, P. B. L.; Fregolente, L. V.; Pinto, G. M F.; Batistella, C. B.; Wolf-Maciel, M. R.; Maciel Filho, R.; Appl. Biochem. Biotechnol. 2008, 146,165.
- 20. Schoenfelder, W.; Eur. J. Lip. Sci. Technol. 2003, 105, 45.
Datas de Publicação
-
Publicação nesta coleção
22 Set 2009 -
Data do Fascículo
2009
Histórico
-
Aceito
20 Jan 2009 -
Recebido
19 Ago 2008