Resumo
In this work the potentiality of photo-Fenton processes were investigated toward the degradation of aromatic hydrocarbons (BTXs) from water contaminated with gasoline. The main results demonstrated that BTXs can be quickly degraded by photo-Fenton process assisted by solar or artificial UV-A radiation, degradation that leads to generation of characteristic phenolic transient species (ie. phenol, hydroquinone and catechol). In the treatment of contaminated water by photo-Fenton processes assisted by solar light, complete BTXs removal was observed in reaction times of about 5 min. Mineralization of about 90% was also observed by applying a multiple H2O2 addition system.
BTXs; photo-Fenton; gasoline-contaminated water
BTXs; photo-Fenton; gasoline-contaminated water
ARTIGO
Degradação de benzeno, tolueno e xilenos em águas contaminadas por gasolina, utilizando-se processos foto-Fenton
Degradation of benzene, toluene and xilenes in gasoline-contaminated waters by photo-Fenton processes
Elaine Regina Lopes TiburtiusI; Patricio Peralta-ZamoraI,* * e-mail: zamora@quimica.ufpr.br ; Alexandre EmmelII
IDepartamento de Química, Universidade Federal do Paraná, CP 19081, 81531-990 Curitiba - PR, Brasil
IICentro Integrado de Tecnologia e educação Profissional, 81310-010 Curitiba - PR, Brasil
ABSTRACT
In this work the potentiality of photo-Fenton processes were investigated toward the degradation of aromatic hydrocarbons (BTXs) from water contaminated with gasoline. The main results demonstrated that BTXs can be quickly degraded by photo-Fenton process assisted by solar or artificial UV-A radiation, degradation that leads to generation of characteristic phenolic transient species (ie. phenol, hydroquinone and catechol). In the treatment of contaminated water by photo-Fenton processes assisted by solar light, complete BTXs removal was observed in reaction times of about 5 min. Mineralization of about 90% was also observed by applying a multiple H2O2 addition system.
Keywords: BTXs; photo-Fenton; gasoline-contaminated water.
INTRODUÇÃO
Episódios de contaminação envolvendo hidrocarbonetos de petróleo são relatados com bastante frequência, principalmente em função dos também frequentes acidentes envolvendo transporte e estocagem de combustíveis.1,2 Nos Estados Unidos, por exemplo, estima-se que 35% dos tanques de armazenamento subterrâneo de gasolina e diesel apresentam problemas de vazamento, o que irremediavelmente resulta na contaminação de solos e águas subterrâneas.3
Dentro deste contexto, destaque deve ser dado a hidrocarbonetos aromáticos como benzeno, tolueno, etilbenzeno e xilenos (coletivamente conhecidos como BTEXs), espécies monoaromáticas de reconhecida toxicidade e elevada solubilidade em água. Em geral, admite-se que os BTEXs representem mais do que 50% da fração de hidrocarbonetos solúveis da gasolina,4 característica que, junto com a sua reduzida adsorção em matrizes de solo,5 favorece a sua mobilização nos sítios contaminados.
Recentemente, a contaminação de águas subterrâneas por combustíveis com elevado teor de álcool tem sido apontada como um problema ambiental emergente,3 principalmente em função de este aditivo facilitar, por efeito cossolvente, a mobilização dos hidrocarbonetos em solos contaminados por derramamento. Em geral, estima-se que a adição de 10% de álcool (etílico ou metílico) favorece largamente a solubilização de benzeno e tolueno em água, com efeitos menos significativos para etilbenzeno e xilenos.3 Trata-se de uma observação que permite prever um potencial poluente ainda maior para a gasolina comercializada no Brasil, a qual é aditivada com cerca de 25% de etanol.
Em função da elevada toxicidade dos BTEXs e da consequente diversidade de efeitos deletérios provocados nos ecossistemas contaminados, a procura por sistemas de remediação apresenta-se extremamente relevante. Neste sentido, é importante salientar que processos de remediação podem ser implementados em três diferentes linhas de atuação. São estas: prevenção de episódios de contaminação; tratamento de solos contaminados e, tratamento de águas contaminadas.
Em função da precária fiscalização, a implementação da primeira alternativa é extremamente desfavorecida, principalmente em países em desenvolvimento. Por sua vez, o tratamento de solos contaminados, utilizando-se barreiras reativas,6 por exemplo, é extremamente dificultado, tanto pela complexidade da matriz de solo, como pela elevada velocidade com que os poluentes solúveis são lixiviados. Assim, embora o tratamento de águas contaminadas represente a última frente de ação, muitas vezes corresponde à única alternativa tecnicamente viável.
Dentro do contexto da remediação de águas contaminadas, e levando-se em consideração o local de tratamento, duas alternativas são possíveis: processos in situ e ex situ. Processos in situ costumam envolver processos de remoção física (ex. air-stripping7) ou, mais frequentemente, processos de bioatenuação natural, fundamentados na ação de micro-organismos autóctones.8 Em geral, os processos físicos permitem uma eficiente remoção dos hidrocarbonetos voláteis. Entretanto, o seu caráter não-destrutivo implica na necessidade de processos auxiliares, orientados a adsorver, degradar ou dispor os hidrocarbonetos previamente extraídos. Por sua vez, os processos de bioatenuação natural caracterizam-se pela extrema morosidade do processo depurativo, mesmo em sistemas estimulados pela adição de nutrientes.8 Adicionalmente, estudos orientados a avaliar a degradação biológica de BTEXs demonstram um significativo efeito inibitório induzido pela presença de etanol, em razão da sua degradação provocar um rápido consumo dos aceptores de elétrons disponíveis no meio.9
Nos processos ex situ, a água é retirada do local contaminado e tratada por processos diversos (sistema pump-and-treat), dentre os que destacam alternativas físicas fundamentadas em adsorção,10 processos biológicos envolvendo fungos11 ou consórcios bacterianos específicos12 e, mais recentemente, processos de oxidação química fundamentados na ação de radical hidroxila (processos oxidativos avançados, POAs).13,14 Dentre os vários POAs estudados como alternativa de tratamento de águas, destaque deve ser dado aos processos foto-Fenton,15 os quais têm demonstrado elevada capacidade de degradação de inúmeros poluentes ambientais, incluindo aqueles sabidamente resistentes frente a outras tecnologias de tratamento. Interessantes revisões relacionadas com o fundamento e as principais aplicações ambientais dos processos Fenton e foto-Fenton podem ser encontradas na literatura especializada, destacando-se o trabalho recentemente publicado por Nogueira e colaboradores.16
Neste trabalho, a potencialidade dos processos Fenton e foto-Fenton foi investigada em relação à remediação de águas contaminadas por gasolina. Várias condições experimentais foram avaliadas, incluindo-se o efeito de fontes artificiais e naturais de radiação.
PARTE EXPERIMENTAL
Reagentes e insumos
Misturas de benzeno (Biotic), tolueno (Merck) e xilenos (Merse), todos de grau analítico PA, foram preparadas em solução aquosa, contendo 20 µg L-1 de cada espécie.
Soluções aquosas de H2O2 (10% m/v) foram preparadas a partir de solução estoque a 50% (m/v), gentilmente fornecida por Peróxidos do Brasil Ltda. A concentração desta solução foi periodicamente avaliada, por titulação permanganométrica.
Como fonte de íons ferrosos foi utilizado sulfato ferroso hepta-hidratado (FeSO4.7H2O, Isofar).
Oxigênio comercial foi adquirido junto à White Martins.
Outros reagentes (ácido sulfúrico Isofar, hidróxido de sódio Vetec, biftalato de potássio Merse, metavanadato de amônio Vetec, reagente de Folin-Ciocalteau Polipur e o-fenantrolina Aldrich) foram de grau analítico PA.
Para avaliar a transferência de substratos entre a fase aquosa e a gasolina uma situação de derramamento foi simulada, utilizando-se condições experimentais similares às reportadas por Ziolli e Jardim.17 Neste ensaio, 4 L de água destilada e 200 mL de gasolina comercial, aditivada com 25% de etanol, foram deixadas em contato passivo durante 48 h, em temperatura de 25 ± 5 ºC. Ensaios foram realizados em recipiente fechado e protegido da luz e em sistema aberto sem nenhum tipo de proteção. Amostras da fase aquosa foram coletadas a cada 12 h, sendo caracterizadas por cromatografia gasosa e espectroscopia de fluorescência molecular.
Tratamento por processos foto-Fenton
O tratamento de soluções aquosas contendo benzeno (substrato padrão), BTXs e águas contaminadas com gasolina foi realizado em sistema aberto, utilizando-se um reator de vidro de 300 mL de capacidade (11 cm de altura e 6 cm de d.i.), equipado com camisa para refrigeração por água, agitação magnética e sistema de oxigenação (vazão média de 45 mL min-1). A radiação foi proporcionada por uma lâmpada a vapor de mercúrio (125 W, Philips), introduzida na solução com a ajuda de um bulbo de quartzo (radiação UV-C) ou de vidro Pyrex (radiação UV-A).
O tratamento envolvendo radiação solar foi realizado em sistema aberto, utilizando-se frascos de vidro borossilicatado de 300 mL de capacidade (11 cm de altura e 6 cm de d.i.), colocados no centro de um coletor parabólico revestido de alumínio. Estudos adicionais foram realizados no modo batelada com recirculação, utilizando-se um reator solar do tipo cilíndrico parabólico composto. O reator é composto por seis tubos de vidro borossilicato de 80 cm de comprimento e 2,6 cm de d.i. (volume iluminado de aproximadamente 2,5 L), um reservatório de amostra com capacidade para 6 L, uma bomba de recirculação e tubos conectores (Figuras 1S, material suplementar). Os refletores foram confeccionados com alumínio, sendo fixados em uma base de madeira com ângulo de inclinação variável, de modo a se obter o máximo de aproveitamento da radiação solar. O volume total tratado foi de 5 L, com vazão de 0,4 L min-1.
Todos os experimentos foram realizados na cidade de Curitiba-PR (25º 25''48's e 49º 16''15'w), em dias claros do mês de março, no horário compreendido entre 10:00 e 13:00 h. Nestas condições, a intensidade média da radiação foi de 3,0 mW cm-2 (300-400 nm), medida em radiômetro Cosmo lux (UVA Test 3000).
Quando sistemas de múltiplas adições de peróxido de hidrogênio foram utilizados, as adições foram realizadas a cada 30 min. Em todos os casos foram adicionadas alíquotas de H2O2 50% (m/v), de maneira a se obter uma concentração de 100 µg L-1.
Em todos os casos, alíquotas foram tomadas em intervalos regulares, filtradas em membrana de acetato de celulose (Milipore, 0,45 µm) e submetidas à análise.
Controles analíticos
A concentração de BTXs foi determinada por cromatografia em fase gasosa, utilizando-se sistema de headspace. As análises foram realizadas em cromatógrafo Varian CP3800, equipado com coluna DB624 e detector de ionização em chama. Curvas analítica foram elaboradas na faixa de concentração compreendida entre 5 e 2000 µg L-1, a partir de um padrão certificado contendo a mistura de benzeno, tolueno, etilbenzeno e xilenos (BTEX) em metanol. Estas curvas foram regularmente verificadas por um padrão e uma carta de controle, aceitando-se um desvio de 15% como limite de intervenção. O limite de quantificação foi estabelecido em 5 µg L-1, enquanto que os coeficientes de variação observados (análise em triplicatas) foram de 1,9; 7,7; 7,6; 10,1 e 12,8%, para benzeno, tolueno, etilbenzeno, m,p-xileno e o-xileno, respectivamente.
Intermediários de degradação de caráter fenólico foram identificados por cromatografia em fase líquida, comparando-se os tempos de retenção observados com os de padrões previamente caracterizados (ex., fenol, benzoquinona, hidroquinona, catecol). As determinações foram realizadas em cromatógrafo Shimadzu LC-10AD, equipado com coluna C18 (hypersil 4,6 mm x 15 cm) e detector ultravioleta SPD-M10A (225 nm). A fase móvel foi constituída de água (Milli-Q) e metanol, na proporção 1:1 (v/v), acidificada com H3PO4 0,01%.
Intermediários de degradação de caráter ácido (ex., ácidos acético, oxálico, fumárico e maleico) foram identificados da mesma forma, utilizando-se uma coluna de troca iônica (Aminex, HPX 87H, 300 x 7,8 mm) e uma fase móvel constituída de H2SO4 8 mmol L-1.
O teor de carbono orgânico total (COT) foi determinado em analisador Shimadzu VCPH TOC. Curvas analíticas foram elaboradas com padrões aquosos de biftalato de potássio.
As concentrações de peróxido de hidrogênio residual, fenois totais e Fe2+/Fe3+ foram determinadas por espectroscopia eletrônica e metodologias fundamentadas na reação com metavanadato de amônio,18 reagente de Folin-Ciocalteau (método APHA-5550B)19 e o-fenantrolina (método APHA 3500D),19 respectivamente. Medidas de absorbância foram realizadas em espectrofotômetro Sinco S-1150, utilizando-se cubetas de quartzo de 1 cm.
RESULTADOS E DISCUSSÃO
Estudos preliminares
Inicialmente, o efeito da concentração de Fe2+ e H2O2 na capacidade de degradação do sistema foto-Fenton foi investigado por um sistema de planejamento fatorial de experimentos, utilizando-se benzeno como substrato padrão. Nesses estudos (resultados não apresentados) foi verificado um forte efeito combinado entre ambas variáveis, assim como uma eficiência de degradação maximizada nas condições experimentais representadas por: pH: 3,0; Fe2+: 10 mg L-1;H2O2: 100 mg L-1. Nestas condições, radical hidroxila é produzido de acordo com a reação apresentada na Equação 1, sendo responsável pela degradação praticamente completa de benzeno em tempos de reação inferiores a 5 min.

Posteriormente, a degradação de soluções aquosas de BTXs foi investigada, utilizando-se as condições previamente definidas e avaliando-se a evolução de formas fenólicas totais. Em geral, admite-se que a degradação de estruturas aromáticas por processos avançados se inicia com a adição de radicais hidroxila, o que leva à formação de estruturas fenólicas características.20 Desta forma, o monitoramento de referido parâmetro pode permitir a apreciação de mecanismos diferenciados, principalmente quando diversas condições experimentais são avaliadas.
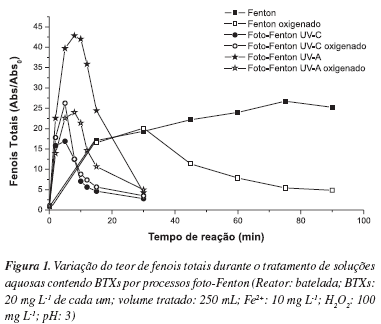
A evolução de formas fenólicas durante o tratamento de soluções aquosas de BTXs, em diferentes condições experimentais, é apresentada na Figura 1. Estes resultados demonstram que os processos assistidos por radiação (UV-C ou UV-A) são cineticamente favorecidos, o que permite um pico de formação de derivados fenólicos nos primeiros minutos de reação (tipicamente 10 min) e a sua completa remoção em maiores tempos de tratamento (aproximadamente 30 min). Trata-se de um comportamento similar ao descrito por Du e colaboradores20 em estudos de degradação envolvendo p-clorofenol. Neste trabalho, o mecanismo de degradação proposto envolve a rápida formação de transientes fenólicos (ex., hidroquinona, benzoquinona e catecol), seguida de reações de abertura de anel e, finalmente, formação de ácidos carboxílicos de cadeia curta (ex., acético, oxálico, fumárico e maleico).
Em contraste, o processo Fenton (não irradiado) leva a uma tardia formação de formas fenólicas, as quais tendem a permanecer até os maiores tempos de reação praticados (90 min). Em função desta observação, o efeito benéfico da radiação é bastante evidente, sendo devido, principalmente, à geração de quantidades adicionais de radical hidroxila, em razão da fotorredução de Fe3+, de acordo com a Equação 2.

Nos processos assistidos por radiação, o efeito da oxigenação não parece decisivo. Nos processos Fenton tradicionais, entretanto, a degradação de BTXs foi consideravelmente favorecida pelo uso de sistemas oxigenados. Embora o papel do oxigênio nos processos avançados ainda seja uma questão em debate,21 estima-se que sua presença permita a geração de espécies ativas de oxigênio, dentre as que destacam oxigênio singlete (1O2), radical superóxido (HO2 ou O2-), além do próprio radical hidroxila (HO).20 Adicionalmente, oxigênio pode reagir com formas radicalares que surgem durante o processo de degradação, formando superóxidos que tendem a se decompor espontaneamente.22
O monitoramento cromatográfico permitiu confirmar a rápida degradação dos BTXs em todas as condições ensaiadas, sendo que, em geral, tratamentos de 5 min permitiram reduzir a concentração de cada um dos substratos em estudo até valores inferiores ao do limite da quantificação (5 µg L-1).
Em função dos mecanismos de reação envolverem reações radicalares, a possibilidade de se formar subprodutos de maior toxicidade que a dos compostos de partida é um fato bem documentado. Em função desta realidade, o monitoramento do processo de mineralização torna-se extremamente importante, de maneira a garantir a completa remoção dos intermediários de reação.23 A evolução do teor de carbono orgânico total (dissolvido) durante o tratamento de soluções aquosas de BTXs por processo foto-Fenton assistido por radiação UV-A é apresentado na Figura 2. A partir destes resultados é possível observar que, na ausência de oxigenação, a adição de apenas uma carga de peróxido induz pequenas taxas de mineralização (menos que 30%), mesmo nos maiores tempos de reação (90 min). A baixa eficiência de mineralização pode ser explicada pela rápida decomposição do H2O2, o que leva a um consumo praticamente completo em tempos de reação de 15 min. A adição sucessiva de peróxido, em intervalos de 30 min, melhora significativamente a capacidade de mineralização do sistema, com remoção máxima da ordem de 90% do teor de COT para sistemas operados com 5 adições consecutivas de peróxido. Este resultado confirma observações realizadas por Oliveira e colaboradores,24 que observaram uma melhor capacidade degradativa dos sistemas Fenton quando H2O2 é adicionado toda vez que a concentração inicial é reduzida em 80%.
Na presença de oxigênio a capacidade degradativa do sistema é significativamente melhorada, o que permite a obtenção de elevadas taxas de mineralização, mesmo com apenas uma adição de peróxido. Em função da perda de BTXs por processos de volatilização (Figura 2), este resultado deve ser analisado com cautela. Entretanto, é importante lembrar que espécies fenólicas são formadas nos primeiros minutos de reação, espécies estas que apresentam uma volatilidade muito inferior à dos compostos de partida. Benzeno, por exemplo, apresenta uma pressão de vapor de 100 mm de Hg a 26 ºC, enquanto que para fenol este parâmetro apresenta valores da ordem de 1 mm de Hg a 40 ºC. Para melhor ilustrar este fato, as perdas de COT provocadas por oxigenação de uma solução aquosa de fenol (20 mg L-1) foram também representadas na Figura 2. O fato das perdas serem praticamente negligenciáveis permite presumir que as perdas de BTXs por volatilização sejam de pouca importância, afetando de maneira pouco significativa a avaliação do processo de mineralização.
Estudos cromatográficos orientados à identificação de intermediários permitiram constatar a presença de fenol, benzoquinona e catecol durante os primeiros 5 min de reação, em sistemas oxigenados ou não. Em maiores tempos de reação, ácido oxálico foi identificado com principal subproduto, em ambas as condições. Trata-se de uma observação coerente com os relatos da literatura, que sugerem a produção de três moléculas de ácido carboxílico por clivagem de benzoquinona.22
Caracterização da água contaminada com gasolina
A gasolina utilizada neste estudo (tipo C) foi previamente caracterizada por cromatografia a gás, constatando-se uma composição coerente com as especificações da Portaria Nº 309 da ANP (27/12/2001). Dentre as principais características é possível salientar um teor de aromáticos da ordem de 14% (m/m) e de componentes oxigenados de aproximadamente 27% (m/m). Em razão do elevado número de constituintes da matriz de gasolina, a determinação de hidrocarbonetos transferidos para a fase aquosa torna-se extremamente complexa. Para facilitar esta avaliação, metodologias fundamentadas em espectroscopia de fluorescência molecular têm sido propostas, principalmente em razão da intensa emissão de alguns importantes hidrocarbonetos aromáticos polinucleares.25
Nestes estudos foi constatada uma rápida transferência de hidrocarbonetos fluorescentes para a fase aquosa, transferência esta que se apresenta diferenciada para os sistemas aberto e fechado. No sistema aberto, sob influência da luz, a transferência se dá mais rapidamente, provavelmente em função da degradação fotoquímica de alguns hidrocarbonetos permitir a formação de espécies de maior solubilidade.26 Adicionalmente, mudanças físicas associadas à evaporação, expansão acompanhada da mudança de densidade, viscosidade e tensão interfacial, tendem a influenciar significativamente o processo de transferência de hidrocarbonetos,26 o que pode explicar as diferenças observadas.
Em função da maior semelhança com situações de derramamento envolvendo tanques subterrâneos, a fase aquosa obtida em sistema fechado foi selecionada para estudos posteriores, sendo mais bem caracterizada por cromatografia gasosa. Nestes resultados (Tabela 1) foram constatadas elevadas concentrações de BTXs, assim como a presença de naftaleno e outros inúmeros hidrocarbonetos aromáticos mono e dissubstituídos. O desaparecimento do etanol do filme de gasolina e o enriquecimento da fase aquosa foram também constatados por cromatografia gasosa. Em geral, etanol se transfere para a fase aquosa com extrema facilidade, o que faz com que, em caso de derramamento, a água funcione como um grande reservatório de etanol no ambiente subterrâneo, viabilizando um aumento na solubilidade dos BTXs e uma drástica redução na eficiência de sistemas de remediação natural.27
Estudos de remediação de águas contaminadas no modo batelada
Nas condições em que o derramamento foi simulado (sistema fechado), a carga orgânica transferida para a fase aquosa, medida em termos de demanda química de oxigênio, foi da ordem de 20.000 mg L-1 de O2. Em função dos processos oxidativos avançados apresentarem uma baixa eficiência de degradação frente a cargas orgânicas desta magnitude,28 as fases aquosas contaminadas foram previamente diluídas, até uma carga orgânica da ordem de 2000 mg L-1 (diluição de 10 vezes). Trata-se de uma operação necessária, uma vez que, além de viabilizar o tratamento por processos avançados, permite ajustar a concentração de hidrocarbonetos a níveis usualmente encontrados em águas contaminadas.29,30
Dentre os vários processos inicialmente avaliados, deu-se preferência àqueles assistidos por radiação UV-A (fontes artificiais e bulbo de vidro) e solar. Nos estudos envolvendo radiação UV-A, o monitoramento por espectrometria de fluorescência indicou a completa remoção dos hidrocarbonetos fluorescentes, em tratamentos de apenas 15 min (Figura 3). Em idêntico tempo de reação, a concentração de BTXs torna-se inferior ao limite da quantificação (5 µg L-1), o que atesta a elevada capacidade do processo na degradação dos substratos em estudo. Nestas condições, a mineralização da carga orgânica foi pouco significativa (aproximadamente 20% de redução do teor de carbono orgânico total em 30 min de reação), provavelmente em razão do já comentado rápido consumo de H2O2.
De maneira geral, estima-se que o principal fator que atenta conta a aplicabilidade dos processos fotoquímicos está representado pelo custo associado ao uso de fontes artificiais de radiação. Deste ponto de vista, a utilização de radiação solar aparece como uma interessante alternativa, barateando custos e viabilizando o desenvolvimento de sistemas orientados ao tratamento em grande escala. Neste trabalho, a eficiência de degradação do processo foto-Fenton foi avaliada no modo batelada, utilizando-se um reator localizado no centro de um coletor parabólico de alumínio. Os resultados (Figura 4) demonstram um perfil de remoção de BTEX bastante similar à do processo assistido por radiação artificial, o que implica na obtenção de concentrações abaixo do limite de quantificação (5 µg L-1) em tratamentos de 5 min.
Utilizando-se espectrometria de fluorescência como ferramenta analítica (resultados não apresentados) foi possível observar um aumento de sinal nos primeiros 30 min de tratamento, assim como um pequeno deslocamento da banda para maiores valores de comprimento de onda, comportamento este que é coerente com a formação de uma espécie intermediária de caráter fenólico. Conforme se avança no tempo de tratamento o sinal de emissão decresce sistematicamente, sendo quase que completamente eliminado em tempos de reação de 90 min.
A remoção de carbono orgânico total apresenta-se pouco significativa (< 30%), principalmente em razão do rápido consumo de peróxido de hidrogênio (Figura 4). Visando melhorar esta taxa de mineralização, um sistema de múltiplas adições de peróxido foi praticado, observando-se os resultados apresentados na Figura 5. De acordo com estes resultados, o efeito da reposição do peróxido é bastante evidente, o que permite a elevação da taxa de mineralização até valores da ordem de 90% (3 adições de peróxido, 90 min de reação).
Uma interessante vantagem adicional apresentada pelo processo foto-Fenton está relacionada com a possibilidade de se utilizar radiação de comprimentos de onda de até 600 nm. Considerando-se que aproximadamente 35% da radiação solar corresponde a esta região espectral, a viabilidade econômica destes processos apresenta-se evidente, principalmente em locais com elevada insolação.31
Estudos de remediação de águas contaminadas no modo batelada com recirculação
Visando ampliar a escala de trabalho, dentro dos limites do trabalho em laboratório, um sistema em batelada com recirculação foi avaliado, utilizando-se um coletor solar do tipo parabólico composto (CPC). Provavelmente este seja um dos coletores solares mais utilizados em estudos deste tipo, pois apresenta a melhor relação óptica para aproveitamento de energia solar. Trata-se de reatores estáticos com uma superfície refletora ao redor do cilindro reator, projetada para aproveitamento da radiação direta e difusa. A radiação refletida é distribuída na parte inferior do tubo, o que faz com que toda a circunferência seja iluminada. Além de proporcionar uma boa eficiência fotônica, o sistema apresenta baixo custo de capital e operação.16,28
A potencialidade deste sistema foi avaliada no tratamento de 2 L de água contaminada, utilizando a remoção do carbono orgânico total como resposta analítica. Os resultados (Figura 6) indicam que um sistema de três adições sucessivas de H2O2 permite uma remoção máxima de COT da ordem de 60%, em tratamentos de 120 min. Trata-se de uma mineralização inferior à observada nos estudos anteriores, provavelmente em razão das diferenças no volume iluminado. De acordo com antecedentes da literatura,32 a geometria dos reatores influencia de maneira decisiva a capacidade de degradação do sistema, principalmente em função das diferenças observadas na intensidade da radiação.
É importante destacar que mesmo após sucessivas adições de peróxido de hidrogênio, concentrações mensuráveis de H2O2 não foram observadas no tempo final de tratamento (120 min).
Eliminação do ferro residual
Embora a concentração utilizada de ferro solúvel (10 mg L-1) seja inferior ao máximo permitido pela Resolução CONAMA 357 para lançamento de resíduos (15 mg L-1), ela excede o valor estipulado para águas doces de classe 3 (5,0 mg L-1). Da mesma forma, esta concentração supera o valor máximo permitido pela Resolução CONAMA 396, em relação ao consumo humano de águas subterrâneas (0,3 mg L-1). Para se enquadrar aos limites impostos pelas resoluções antes mencionadas, duas alternativas são possíveis. A primeira consiste na remoção do ferro solúvel por precipitação em meio alcalino seguida de floculação, como proposto por El-Gohary e colaboradores,33 enquanto que a segunda envolve a aplicação de processos Fenton com formas imobilizadas de ferro. Recentemente, a completa degradação de hidroquinona por processos foto-Fenton envolvendo o uso de ferro imobilizado em fibras de sílica foi relatada.34 Neste estudo, concentrações de ferro solúvel inferiores a 1,2 mg L-1 foram observadas, o que atesta a viabilidade deste tipo de propostas.
CONCLUSÕES
Os resultados anteriormente relatados demonstram a potencialidade dos processos foto-Fenton, em relação à remediação de águas contaminadas com hidrocarbonetos de petróleo.
Processos assistidos por radiação (UV-C, UV-A e solar) permitem uma rápida degradação dos BTXs, o que leva à sua completa remoção em tempos de reação da ordem de 5 min. A referida degradação envolve a formação de transientes fenólicos, os quais são também completamente degradados em maiores tempos de reação (tipicamente 30 min). Para mineralização da carga orgânica condições mais vigorosas são necessárias, destacando-se a reposição do peróxido de hidrogênio em intervalos de 30 min.
Além de uma elevada eficiência de degradação, o sistema apresenta uma marcante simplicidade operacional, principalmente em função do seu caráter homogêneo. Dentro deste contexto, destaque deve ser dado aos sistemas assistidos por radiação solar, os quais viabilizam uma significativa redução do custo de tratamento.
MATERIAL SUPLEMENTAR
Encontra-se disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre. Na Figura 1S apresentam-se fotografias do reator utilizado nos estudos de degradação realizados no modo batelada com recirculação.
Recebido em 15/9/08; aceito em 24/4/09; publicado na web em 30/9/09
MATERIAL SUPLEMENTAR

Figura 1S - Clique para ampliar
- 1. Farhadian, M.; Duchez, D.; Vachelard, C.; Larroche, C.; Water Res. 2008, 42, 1325.
- 2. De Nardi, I. R.; Ribeiro, R.; Zaiat, M.; Foresti, E.; Process Biochem. 2005, 40, 587.
- 3. Lee, K.Y.; Environ. Pollut. 2008, 154, 320.
- 4. Kermanshahi, A.; Karamanev, D.; Margaritis, A.; Biochem. Eng. J. 2006, 27, 204.
- 5. Langwaldt, J. H.; Puhakka, J. A.; Environ. Pollut. 2000, 107, 187.
- 6. Vesela, L.; Nemecek, J.; Siglova, M.; Kubal, M.; Int. Biodeterior. Biodegrad. 2006, 58, 224.
- 7. Ruey-Shin, J.; Su-Hsia, L.; Min-Chih, Y.; J. Membr. Sci. 2005, 255, 79.
- 8. Farhadian, M.; Vachelard, C.; Duchez, D.; Larroche, C.; Bioresour. Technol. 2008, 99, 5293.
- 9. Lovanh, N.; Hunt, C. S.; Alvarez, P. J. J.; Water Res. 2002, 36, 3739.
- 10. Lu, C.; Su, F.; Hu, S.; App. Surf. Sci. 2008, 254, 7035.
- 11. García-Peña, I.; Ortiz, I.; Hernández, S.; Revah, S.; Int. Biodeterior. Biodegrad. 2008, no prelo.
- 12. Littlejohns, J. V.; Daugulis, A. J.; Process Biochem. 2008, no prelo.
- 13. Mascolo, G.; Ciannarella, R.; Balest, L.; Lopez, A.; J. Hazard. Mater. 2008, 152, 1138.
- 14. Schierz, G. A.; Trommler, U.; Horwitz, C. P.; Collins, T. J.; Kopinke, F. D.; Appl. Catal. B-Environ. 2007, 72, 26.
- 15. Neyens, E.; Baeyens, J.; J. Hazard. Mater. 2003, 98, 33.
- 16. Nogueira, R. F. P.; Trovó, A. G.; da Silva, R. A.; Villa, R. D. de Oliveira, M. C.; Quim. Nova 2007, 30, 400.
- 17. Ziolli, R. L.; Jardim, W. F.; J. Photochem. Photobiol. A-Chem. 2002, 147, 205.
- 18. Oliveira, M.C.; Nogueira, R. F. P.; Neto, J. A. G.; Quim. Nova 2001, 24, 188.
- 19. APHA; Standard Methods for Examination of Water and Wastewater, 19th ed., APHA: New York, 1995.
- 20. Du, Y.; Zhou, M.; Lei, L.; J. Hazard. Mater. 2007, 139, 108.
- 21. Zhang, D.; Qiu, R.; Song, L.; Eric, B.; Mo, Y.; Huang, X.; J. Hazard. Mater. 2008, no prelo.
- 22. Du, Y.; Zhou, M.; Lei, L.; Water Res. 2007, 41, 1121.
- 23. Robert, D.; Malato, S.; Sci. Total Environ. 2002, 291, 85.
- 24. Oliveira, I. S.; Viana, L.; Verona, C.; Fallavena, V. L. V.; Azevedo, C. M. N.; Pires, M.; J. Hazard. Mater. 2007, 146, 564.
- 25. Abbas, O.; Réfuba, C.; Dupuy, N.; Permanyer, A.; Kister, J.; Azevedo, D.; Fuel 2006, 85, 2653.
- 26. Ganjali, S. T.; Niknafs, B. N.; Khosravi, M.; Iranian J. Environ. Health Sci. Eng. 2007, 4, 37.
- 27. Kaipper, A. I. B.; Tese de Doutorado, Universidade Federal de Santa Catarina, Brasil, 2003.
- 28. Malato, S.; Blanco, J.; Vidal, A.; Richter, C.; Appl. Catal. B-Environ. 2002, 37, 1.
- 29. Rosell, M.; Lacorte, S.; Ginebreda, A.; Barceló, D.; J. Chromatogr., A 2003, 995, 171.
- 30. Garoma T.; Gurol, M. D.; Osibodu, O.; Thotakura, L.; Chemosphere 2008, no prelo.
- 31. Rincón, A. G.; Pulgarin, C.; Catal. Today 2007, 124, 204.
- 32. Gernjak, W.; Krutzler, T.; Glaser, A.; Malatao, S.; Caceres, J.; Bauer, R.; Fernandez-Alba, A.R.; Chemosphere 2003, 50, 71.
- 33. El-Gohary, F. A.; Badawy, M. I.; El-Khateeb, M. A; El-Kalliny, A. S.; J. Hazard. Mater. 2009, 162, 1536.
- 34. Moncayo-Lasso, A.; Torres-Palma, R. A.; Kiwi, J.; Benítez, N.; Pulgarin, C.; Appl. Catal. B-Environ. 2008, 84, 577.
Datas de Publicação
-
Publicação nesta coleção
26 Nov 2009 -
Data do Fascículo
2009
Histórico
-
Aceito
24 Abr 2009 -
Recebido
15 Set 2008