Resumo
This paper proposes a methodology for spectrophotometric determination of hexamethylenetetramine (HMT) by using chromotropic acid in a phosphoric acid media employing a domestic microwave oven as a source of heating. The reddish-purple soluble product is quantitatively formed after 30 s of irradiation and obeys the Beer´s law in the range between 0.1-1.2 mg L-1 HMT (r = 0.99925). The method was applied successfully in commercial pharmaceutical preparations containing dyes in their composition. The results showed that the method proposed is feasible for simplicity, speed, low cost, precision and accuracy when compared with United States Pharmacopeia official method.
hexamethylenetetramine; chromotropic acid; microwave oven
hexamethylenetetramine; chromotropic acid; microwave oven
NOTA TÉCNICA
Determinação espectrofotométrica de hexametilenotetramina (HMT) em medicamentos, utilizando ácido cromotrópico e forno de micro-ondas
Spectrophotometric determination of hexamethylenetetramine (HMT) in pharmaceutical preparations using chromotropic acid and microwave oven
Enelton FagnaniI, * * e-mail: enelton@fec.unicamp.br ; Leonardo PezzaII; Helena Redigolo PezzaII
IFaculdade de Engenharia Civil, Arquitetura e Urbanismo, Universidade Estadual de Campinas, CP 6021, 13083-852 Campinas - SP, Brasil
IIInstituto de Química, Universidade Estadual Paulista, CP 355, 14801-970 Araraquara - SP, Brasil
ABSTRACT
This paper proposes a methodology for spectrophotometric determination of hexamethylenetetramine (HMT) by using chromotropic acid in a phosphoric acid media employing a domestic microwave oven as a source of heating. The reddish-purple soluble product is quantitatively formed after 30 s of irradiation and obeys the Beer´s law in the range between 0.1-1.2 mg L-1 HMT (r = 0.99925). The method was applied successfully in commercial pharmaceutical preparations containing dyes in their composition. The results showed that the method proposed is feasible for simplicity, speed, low cost, precision and accuracy when compared with United States Pharmacopeia official method.
Keywords: hexamethylenetetramine; chromotropic acid; microwave oven.
INTRODUÇÃO
A hexametilenotetramina (HMT, 1,3,5,7-tetrazociclotetradecano, metenamina, urotropina, aminofórmio) vem sendo utilizada como importante antisséptico do trato urinário desde 1894, quando Nicolaier percebeu que a administração oral desse composto levava à liberação do formaldeído em contato com os ácidos urinários.1 Mesmo atuando como germicida, a HMT já pode, segundo a literatura, ser biodegradada por ação fúngica em sistemas de tratamento de esgoto por lodo ativado.2 Além de sua importância farmacêutica, a HMT também é muito utilizada na composição de explosivos3,4 e resinas.5 Sua estrutura molecular pode ser vista na Figura 1.
De acordo com a literatura,6 a hidrólise ácida de HMT libera quantitativamente os produtos amônio e formaldeído, segundo a reação:

A quantificação do formaldeído liberado na reação pode ser uma alternativa viável para a determinação indireta de HMT.
Várias técnicas e metodologias podem ser utilizadas na quantificação de formaldeído7 como, por exemplo, titulometria,8 condutometria,9 potenciometria,10 cromatografia líquida de alta eficiência,11 espectrofluorimetria,12 derivação em fluido supercrítico,13 espectrometria de massas14 e espectrofotometria na região do visível.15,16
Os métodos titulométricos são bastante simples de serem executados, mas têm como característica negativa a baixa seletividade. Os métodos cromatográficos são bastante utilizados, mas atestam contra si o fato de requererem analistas bem treinados, serem onerosos em termos instrumentais e usarem solventes orgânicos, gerando resíduos químicos que merecem atenção especial no descarte. Alternativa intermediária seria o uso da espectrofotometria, com destaque para o método do ácido cromotrópico (ACT), o qual apresenta rapidez, simplicidade, precisão, repetibilidade, reprodutibilidade e baixo custo relativo, quando comparado aos demais procedimentos existentes na literatura.
O método do ácido cromotrópico já era estudado desde Feigl,17 mas foram Georghiou e Ho18 que propuseram o mecanismo de reação aceito até os dias atuais, onde duas moléculas do ácido cromotrópico (ACT) são unidas quando reagem com uma de formaldeído, em meio fortemente ácido (H2SO4) e sob aquecimento em banho, através de uma ponte metilênica. Esta estrutura intermediária é desidratada numa etapa seguinte, na posição dos grupos (-OH) fenólicos, formando-se um heterociclo. Por último, uma etapa de oxidação produz a estrutura colorida de interesse analítico, um composto dibenzoxantílico monocatiônico, planar e aromático, responsável por uma banda de absorção da luz visível na região entre 570-580 nm. O mecanismo proposto para esta reação pode ser visto na Figura 2.
A estrutura marcada como (I) na Figura 2 corresponde à molécula de ACT, enquanto que (IV) corresponde ao produto final púrpura avermelhado. Este composto pode, então, ser usado nas quantificações de formaldeído ou de compostos que o liberem via alguma reação química, como é o caso da HMT.
Apesar de clássica, esta metodologia de análise de formaldeído ainda é bastante estudada, sendo propostas otimizações para o método, como a substituição do ácido sulfúrico por outros menos corrosivos, a elucidação de etapas do mecanismo de formação do composto colorido final19 e a utilização da irradiação de micro-ondas como fonte de aquecimento,20 eliminando-se a morosa etapa de desenvolvimento de cor usando-se banho-maria, por um tempo de 60 min, de acordo com o método original ainda hoje recomendado pelo National Institute for Occupational Safety and Health (NIOSH).15
Essas últimas contribuições da literatura foram fundamentais para a elaboração do presente estudo, onde se propõe um método espectrofotométrico para a determinação de HMT utilizando-se ácido cromotrópico (ACT) em meio a ácido fosfórico, sob irradiação em forno de micro-ondas doméstico por 30 s. A Lei de Lambert-Beer é seguida entre 0,1 e 1,2 mg L-1, na região da luz visível (575 nm) e amostras de medicamentos contendo corantes em sua formulação, onde a HMT é o princípio ativo, foram analisadas com sucesso utilizando-se prévia separação destes corantes.21
PARTE EXPERIMENTAL
Instrumentação
As leituras de absorbância a 575 nm foram realizadas em espectrofotômetro Hewlett Packard HP8453, com cela de 1 cm de caminho óptico. As tomadas de volume foram feitas com vidraria grau "A", além de micropipetas Brand e Eppendorf. A irradiação de micro-ondas foi feita em forno doméstico Panasonic Junior Plus Intelligent Chaos 1100 W. A homogeneização das amostras diluídas de fármacos foi obtida com o auxílio de um banho de ultrassom Thornton T14 com temporizador. Medidas de oxigênio dissolvido foram realizadas com um oxímetro Hanna Instruments HI 9142 e a vazão dos gases de purga foi monitorada por intermédio de um fluxímetro Gilmont Instruments GF 2200HR, nº 12. Colunas para a extração de corantes foram montadas em seringas plásticas Becton & Dickinson (BD) com capacidade de 25 mL, recheadas de algodão hidrófilo, em formato de bolas.
Reagentes e materiais
Foram utilizados reagentes de pureza analítica e todas as soluções foram preparadas com água deionizada: Formaldeído 37%, Mallinckrodt - preparou-se por diluição uma solução com concentração de 1000 mg L-1. Essa solução foi padronizada e armazenada em local fresco e ao abrigo de luz, mantendo suas características por até 3 meses; Ácido fosfórico concentrado 85%, Mallinckrodt - estabeleceu-se um fluxo de 200 mL min-1 de oxigênio gasoso medicinal por 40 min e saturou-se 500 mL do reagente neste gás; Água de preparo das soluções - saturada em oxigênio por borbulhamento do gás medicinal, sob as mesmas condições usadas para o ácido fosfórico; Ácido cromotrópico, sal dissódico di-hidratado (C10H8O8S2.2Na.2H2O), (pureza > 90%) Merck - preparou-se diariamente uma solução a 10% (m V-1); Hexametilenotetramina (C6H12N4) 99,9% pura, Merck - sólido seco sob pentóxido de fósforo por 4 h e conservado em dessecador com cloreto de cálcio anidro. Preparou-se diariamente solução 100 mg L-1 para a construção da curva analítica.
Calibração do forno de micro-ondas doméstico
A potência das micro-ondas que são emitidas pelo magnetron de um forno doméstico está normalmente entre 600-700 W. Pode-se obter um valor mais preciso, medindo-se o aumento de temperatura em uma quantidade de água tal que absorva essencialmente toda a energia dispensada à cavidade do forno.
A calibração da potência efetiva do forno de micro-ondas doméstico foi adaptada da literatura,22, como segue: adicionou-se 1 L de água deionizada a um béquer de vidro, posicionando-o no centro do prato do forno de micro-ondas. Mediu-se a temperatura inicial com um termômetro e, em seguida, submeteu-se a água a aquecimento durante 2 min à potência alta. Ao final do aquecimento, mediu-se a temperatura final com o mesmo termômetro e procedeu-se o cálculo da potência absorvida pela amostra, em Watts, utilizando-se a seguinte fórmula:

onde: P = potência aparente absorvida pelo fluido (W); Cp = capacidade calorífica do fluido (cal grau-1); K = fator de conversão (calorias para Watts); ΔT = variação de temperatura (ºC); m = massa do fluido (g); t = tempo (s).
Quando se utiliza água, essa fórmula se reduz para P = 35 x ΔT.
Estudo do tempo de irradiação do sistema
Diversos ensaios foram realizados, variando-se o tempo de irradiação sobre a mistura reacional em forno de micro-ondas doméstico, na potência máxima, observando-se qual deles seria o mais eficiente, ou seja, apresentando melhor sinal analítico e sem perdas em repetibilidade.
Para tal, em um béquer de 50 mL, posicionado sobre uma placa de Petri, colocada no centro do prato do forno de micro-ondas, adicionaram-se 0,5 mL de uma solução de HMT 40 mg L-1, 5,0 mL de ácido fosfórico concentrado (H3PO4) e 0,5 mL de solução 10% (m V-1) de ACT. Cobriu-se o béquer com vidro de relógio e submeteu-se a mistura à irradiação de micro-ondas, em intervalos de 5, 10, 15, 20, 25, 30 ou 35 s, em triplicata. Após cada intervalo e respectivo esfriamento em temperatura ambiente, transferiu-se e avolumou-se com água deionizada, em balão volumétrico de 25 mL, procedendo-se à leitura de absorbância em 575 nm.
Concentração de ACT utilizada
Otimizou-se a concentração de ácido cromotrópico por meio de alguns experimentos, realizados em triplicata, conforme as condições descritas na Tabela 1. Partiu-se de uma solução de ACT a 20%, diluindo-se adequadamente com água deionizada para se chegar às concentrações finais de 5 e 10%, num volume adicionado de 0,500 mL. Brancos analíticos foram analisados respectivamente para cada teste, utilizando-se água deionizada em lugar de HMT. Após as amostras esfriarem à temperatura ambiente, foram transferidas para balão volumétrico de 25 mL, avolumadas com água deionizada e os valores de absorbância a 575 nm foram então medidos.
Comparou-se o resultado obtido por Gigante, Pezza e Pezza,23 no qual o método do ácido cromotrópico foi usado para quantificar um composto liberador de formaldeído via reação química apropriada.
Quantidade utilizada de H3PO4 concentrado
Definido o tempo máximo de aquecimento em forno de micro-ondas permitido para a mistura, sem que houvesse perdas em repetibilidade, partiu-se para a determinação do volume ótimo para se trabalhar com ácido fosfórico.
Na Tabela 2 pode-se ver as condições utilizadas para testar o efeito do volume de ácido fosfórico concentrado utilizado na reação entre HMT e ACT 10% em forno de micro-ondas.
Os testes foram realizados em triplicata e brancos analíticos foram feitos nas mesmas condições, utilizando-se água deionizada no lugar de HMT. Transferiram-se as soluções para balões volumétricos de 25 mL e realizou-se a leitura de absorbância, a exemplo do que foi feito no item anterior.
Estabilidade óptica do produto formado
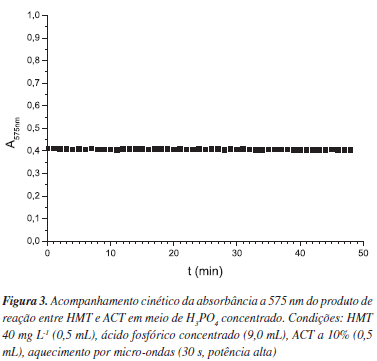
Procedeu-se um estudo da estabilidade óptica do produto formado, utilizando-se as mesmas condições descritas no estudo do tempo de irradiação do sistema. Submeteu-se essa solução então a leituras espectrofotométricas a 575 nm, minuto a minuto por 48 min. Realizou-se ainda uma leitura isolada após 48 h. Esse procedimento foi realizado em triplicata.
Influência da oxigenação do meio
Segundo a literatura,19 a reação entre CH2O e ACT em meio fortemente ácido e sob aquecimento é bastante dependente da oxigenação do meio líquido, quando outro ácido, que não o sulfúrico concentrado, é utilizado como reagente. Conforme esquematizado na Figura 2, a última etapa da reação envolve uma oxidação que, ainda segundo a literatura,19 se realiza plenamente na presença de ácido sulfúrico concentrado, mas que é parcial na presença de ácido sulfúrico diluído a 50% ou quando se utiliza o ácido clorídrico ou fosfórico, mesmo concentrados.
Para verificar a afirmação, no caso da reação com HMT, curvas analíticas comparativas foram construídas, utilizando-se reagentes com e sem saturação em oxigênio, no preparo dos padrões e do branco analítico, com o intuito de observar este efeito sobre a absorbância. No caso do teste com o meio rico em O2, utilizou-se água deionizada (para o preparo dos reagentes) e um ácido fosfórico concentrado recém- saturados, via borbulhamento do gás medicinal. A partir de uma solução 100 mg L-1 de HMT, ACT 10% (m V-1) e H3PO4 concentrado, efetuou-se o planejamento das curvas analíticas.
Após desenvolvimento de cor, as soluções foram quantitativamente transferidas para balões volumétricos de 25 mL, completando-se o volume com água deionizada e procedendo-se às respectivas leituras de absorbâncias a 575 nm.
Análise dos medicamentos comerciais
Três medicamentos de circulação nacional contendo HMT como princípio ativo,24 além de corantes diversos, como vermelho bordeau, vermelho ponceau, azul de metileno e acriflavina, foram analisados em triplicata. Os medicamentos estudados foram de diferentes fabricantes, com formulações e quantidades de princípio ativo também diferentes, mas usados para o mesmo tipo de enfermidade: infecções do trato urinário. Entre 5 e 8 drágeas de cada medicamento foram maceradas em almofariz de ágata e, homogeneizado o pó, pesou-se uma massa correspondente à média das massas das drágeas, em cada caso, e transferiu-se quantitativamente para balão volumétrico de 500 mL, no caso do medicamento Cystex (250 mg HMT, nominal vide bula) ou de 250 mL no caso dos demais (120 mg HMT, nominal vide bula), avolumando-se com água deionizada. Devido à presença de corantes e alguns excipientes nas drágeas foi necessário realizar a separação das substâncias. Para tanto, submeteu-se 25 mL da solução ao banho ultrassom por 15 min, seguido de eluição em coluna de algodão como proposto na literatura.21 Uma seringa plástica com capacidade de 25 mL foi carregada com 3 bolas de algodão hidrófilo (aproximadamente 3,5 g), ocupando aproximadamente 2/3 do seu volume. Cuidadosamente, adicionou-se a amostra à coluna deixando-se a eluição seguir por gravidade. Após esse tratamento prévio, 500 µL desse eluato foram tomados e submetidos ao mesmo procedimento e condições dadas aos padrões da curva analítica, como já descrito acima.
Validação da metodologia proposta
O método proposto foi comparado ao descrito pela Farmacopéia Americana USP,25 o qual utiliza a titulometria ácido-base por retorno, modificado da seguinte forma: uma certa massa de comprimidos macerados, contendo teoricamente 100 mg de HMT, foi diluída em 35 mL de água deionizada, sonificada por 15 min e transferida quantitativamente para balão volumétrico de 50 mL. Uma alíquota de 10,00 mL foi transferida do balão para um erlenmeyer de 250 mL, onde se adicionaram 25,00 mL de uma solução padronizada de H2SO4 0,500 mol L-1 e se aqueceu a mistura em chapa a 90 ºC até que todo o formaldeído fosse liberado (comprova-se por teste qualitativo utilizando solução de ACT 5% e H2SO4 concentrado). Após esfriar a mistura até a temperatura ambiente, procedeu-se à titulação do excesso de ácido restante, utilizando-se solução padronizada de NaOH 0,800 mol L-1. Devido à presença dos corantes nas formulações farmacêuticas, valeu-se de um eletrodo de vidro e de titulador automático para se realizar a determinação volumétrica, já que neste caso não é possível utilizar o indicador visual fenolftaleína como preconiza o Método Oficial.25
RESULTADOS E DISCUSSÃO
Apesar da potência nominal do forno de micro-ondas utilizado ser de 1100 W, após procedimento de calibração chegou-se a um resultado efetivo menor, em torno de 675 W, que está dentro da faixa normalmente encontrada na literatura (600-700 W).22 Fatores como dissipação dielétrica e perdas radiantes são os responsáveis por essa diferença. Características da matriz e a temperatura inicial de trabalho influem nestas perdas de potência efetiva, sendo recomendado trabalhar sempre na mesma temperatura inicial (20 ± 1 ºC). O uso de micro-ondas como fonte de aquecimento diminuiu consideravelmente o tempo de análise (de 1 h em banho-maria15 para 30 s em forno de micro-ondas doméstico) devido ao efeito catalítico deste tipo de radiação eletromagnética, capaz de orientar os dipolos das moléculas, tornando os choques entre elas melhor orientados, aumentando a eficiência de transformação dos reagentes em produtos e diminuindo a energia de ativação do sistema. No estudo do tempo de irradiação, verificou-se que períodos inferiores a 15 s foram insuficientes para o total desenvolvimento de cor da reação, ao passo que o tempo de 35 s levou à maior absorbância, mas sem repetibilidade, já que não raro ocorria o enegrecimento da mistura reacional, resultado de carbonização, além de ebulição com projeção da solução para fora do béquer, concomitantemente à quebra do vidro de relógio ou mesmo do béquer de reação. Adotou-se, portanto, o tempo de irradiação igual a 30 s.
O produto colorido formado na reação entre HMT e ACT, em meio de ácido fosfórico concentrado e aquecimento por micro-ondas, apresentou as mesmas características espectrais que o obtido pela reação entre CH2O e ACT, em meio de ácido sulfúrico concentrado e aquecimento em banho-maria,15 o que indica se tratar, então, do mesmo composto. As condições aqui descritas para se quantificar a HMT foram suficientes para promover a sua hidrólise, a qual libera formaldeído e, na sequência, promover a reação entre este formaldeído liberado e ACT, resultando no composto colorido dibenzoxantílico monocatiônico descrito por Georghiou e Ho.18
Os estudos de otimização da concentração de ACT e do volume de H3PO4 resultaram em valores de absorbância que foram comparados, no intuito de se determinar as melhores condições para a reação. Obtiveram-se valores médios com desvios padrão da ordem de 5%. Os resultados podem ser conferidos nas Tabelas 3 e 4.
Pelos experimentos realizados para a concentração de ACT, estimou-se que a concentração ótima deste reagente é 10%, em acordo com o valor igualmente obtido por Gigante e colaboradores.23 Os valores de absorbância entre as concentrações de 10 e 20% foram bastante próximos, portanto, optou-se por aquela que consumia menos reagente, a 10%.
Variou-se o volume de H3PO4 concentrado utilizado em 5,0; 7,0; 8,0; 9,0 e 10,0 mL, sendo que o valor de 9,0 mL foi aquele que apresentou maior absorbância média. A partir desse valor, a absorbância começa a apresentar uma queda no valor, provavelmente por problemas de eficiência no aquecimento.
O teste de acompanhamento cinético da estabilidade óptica do produto (Figura 3) mostrou que a absorbância obtida foi invariável durante 48 min de medição. Medidas isoladas após 48 h (dados não apresentados) comprovaram que o sinal analítico permanece o mesmo, dentro de uma variação da ordem de 1%, em todas as três replicatas realizadas.
A solução de ACT, desde que guardada em frasco fechado e ao abrigo de luz, é estável por várias horas. Quando degradada, o efeito é visual, pois se percebe um escurecimento da solução originalmente amarela para marrom, tornando fácil a identificação de inadequabilidade do reagente, quando for o caso.
A remoção dos corantes presentes nas formulações estudadas, como azul de metileno, vermelho bordeau, vermelho ponceau e acriflavina, foi bastante eficaz, clarificando a amostra sem, contudo, reter o princípio ativo de interesse, conforme testes de recuperação realizados utilizando-se soluções puras de HMT e soluções dos fármacos analisados. Dentro da faixa de concentração utilizada para as curvas analíticas, a variação nos testes de recuperação para HMT foi em torno de 0,5%, perfeitamente aceitável dentro do erro admitido para determinações de fármacos em drágeas, que é de 5% segundo a Farmacopéia Americana.25 Pode-se ver na Figura 4 os espectros na faixa UV-visível dos corantes normalmente encontrados nos fármacos estudados e fica perceptível a necessidade de sua eliminação, principalmente do azul de metileno, para leituras a 575 nm.
O método do ácido cromotrópico é altamente seletivo para formaldeído,7 não desenvolvendo cor para os demais aldeídos de cadeia maior. Portanto, esses não configuram fonte de interferência analítica. Outros potenciais interferentes, como nitrito, nitrato, acroleína, fenol e cátions de metais de transição como o cromo, não se fazem presentes nas amostras de fármacos estudadas.
A possibilidade de se usar ácido fosfórico ao invés de ácido sulfúrico foi comprovada na literatura19 e é uma vantagem interessante do método aqui proposto sobre o método tradicional, já que o fosfórico é um ácido muito mais seguro de se manusear e menos corrosivo, comparativamente ao sulfúrico. Na Figura 5, pode-se ver a necessidade em se saturar o meio líquido com O2, quando se trabalha com um ácido que não é o sulfúrico, para se conseguir uma faixa mais ampla de linearidade.
Quando o meio foi saturado em oxigênio gasoso, verificou-se linearidade da curva analítica para HMT no intervalo de 0,1 a 1,2 mg L-1 (7,13x10-7 mol L-1 - 8,56x10-6 mol L-1), com um coeficiente de correlação (r) igual a 0,99925. O limite de detecção (LD) calculado segundo a recomendação de Miller & Miller26 foi de 0,016 mg L-1 (1,14x10-7 mol L-1). Em meio não saturado, obtém-se linearidade somente até 0,6 mg L-1 HMT. Após essa concentração, desvios negativos da lei de Lambert-Beer são observados, pela limitação da capacidade oxidante do meio, fundamental à formação do composto colorido de interesse, Figura 2, na transição da estrutura (III) para a estrutura (IV).
Comparativamente à literatura, as potencialidades analíticas do método são bastante aceitáveis e promissoras, já que os artigos publicados mais recentemente para a quantificação de HMT em formulações farmacêuticas apresentam limites de detecção maiores do que o método aqui proposto, faixas de trabalho geralmente mais elevadas e ainda necessitam utilizar solventes orgânicos.27 Os mais recentes estudos apresentaram faixas de trabalho em HMT que variaram de 2,0-35,0 mg L-1 por espectrofotometria,27 0,6-40 mg L-1 por fluorimetria27 e 35,0-7000 mg L-1 por cromatografia líquida de alta eficiência.28 Quando se leva em conta o limite inferior da faixa linear, chega-se à conclusão de que o método aqui proposto apresenta um valor de 20, 6 e 350 vezes mais baixo para quantificação de HMT, respectivamente, mostrando maior sensibilidade do que os demais.27,28 Apesar de frequentemente as metodologias envolvendo cromatografia líquida apresentarem uma melhor sensibilidade quando comparadas às espectrofotométricas, não é o caso quando o analito é HMT.
Na amostragem das drágeas para análise, verificou-se que as formulações apresentam uma variação em massa entre 3 e 4% comparativamente ao valor médio calculado. Essas diferenças podem ser vistas na Tabela 5 e já sinalizam uma fonte de erro em potencial.
Os resultados obtidos na análise dos medicamentos podem ser visualizados na Tabela 6.
Com base nos resultados da Tabela 6, pode-se afirmar que estes mostram boa concordância entre o método oficial25 adaptado e o método proposto, apresentando assim precisão e exatidão comparáveis. Realizando teste t de Student, obteve-se valor de t = -0,1996 e p = 0,9103, em nível de significância de 95%, evidenciando não haver diferença significativa para os resultados obtidos em ambos os métodos para os três produtos farmacêuticos estudados.28
Uma comparação pode ser feita ainda entre a metodologia apresentada por Tininis e colaboradores21 e a proposta neste trabalho. A primeira propõe uma metodologia baseada na titulação ácido-base por retorno, conforme preconizado pela Farmacopéia Americana, introduzindo uma etapa de remoção de corantes, o que possibilita o uso de indicadores visuais (como a fenolftaleína) no procedimento.
A metodologia aqui proposta parte do princípio de que, apesar da titulometria ácido-base ser uma ferramenta importante e apresentar facilidade operacional com baixo custo, ao mesmo tempo se mostra sujeita a certos erros analíticos aos quais a espectrofotometria com ACT não é susceptível, dada sua alta especificidade.
Acredita-se, portanto, que as duas metodologias são eficientes na determinação de HMT em formulações farmacêuticas, na forma como foram apresentadas. Porém, para atender a uma demanda onde a concentração do princípio ativo necessite ser determinada em quantidades traço, ou ainda, em matrizes mais complexas como fluidos biológicos, a metodologia colorimétrica tem maior probabilidade de ser adaptada com sucesso. Além disso, os aspectos positivos da titulometria, como simplicidade, rapidez e baixo custo, seriam igualmente logrados com a espectrofotometria. Portanto, o intuito de se apresentar essa nova metodologia foi aplicar analiticamente os conhecimentos adquiridos pelos estudos do mecanismo da reação entre CH2O e ACT, obtidos por Fagnani e colaboradores,19 além de contribuir com a literatura, apresentando novos caminhos para solucionar novos problemas que venham a surgir.
Normalmente, as metodologias analíticas usadas para fins ambientais são adaptações de análises usadas em alimentos ou em microbiologia clínica. Assim sendo, uma metodologia que investigue um analito com essas características, como é o caso do formaldeído, mas que apresente menor susceptibilidade às interferências, rapidez e baixo custo, é sempre bem-vinda para ser devidamente adaptada e utilizada em análises de impacto antropogênico e lançamento de efluentes, seja em rede coletora ou diretamente no corpo d´água, entre outros.
CONCLUSÃO
O método oficial para análise de formaldeído15 utiliza, além do ACT, ácido sulfúrico concentrado e aquecimento em banho-maria por 1 h, em tubos de ensaio. Este estudo avaliou a possibilidade de se utilizar ácido fosfórico ao invés do sulfúrico, e aquecimento por alguns segundos em forno de micro-ondas doméstico, em substituição ao moroso aquecimento em banho por 1 h.
Além da simplicidade, o método proposto utiliza reagentes baratos e facilmente disponíveis no mercado já em grau de pureza adequado, não necessitando de procedimentos adicionais de purificação. Pela análise dos resultados apresentados, pode-se concluir que a metodologia proposta pode perfeitamente ser utilizada na determinação espectrofotométrica de hexametilenotetramina presente em formulações farmacêuticas que contenham substâncias coloridas, apresentando repetibilidade, sensibilidade e baixo custo, além de uma seletividade maior do que o método titulométrico oficial. A variação dos resultados ficou dentro da faixa dos 5%, que é o preconizado pela Farmacopéia Americana para este tipo de formulação.
AGRADECIMENTOS
À FAPESP e CAPES pelo suporte financeiro.
REFERÊNCIAS
1. Nickel, J. C.; J. Urol. 2005, 173, 21.
2. Kaszycki, P.; Koloczek, H.; Biodegradation 2002, 13, 91.
3. Kirk, R. E.; Othmer, D .F.; Encyclopedia of chemical technology, The Interscience Encyclopedia: New York, 1951.
4. Sharma, S. P.; Lahiri, S. C.; J. Indian Chem. Soc. 2008, 85, 273.
5. Turi, E. A.; Thermal caracterization of polymeric materials, Academic Press: Orlando, 1981.
6. Madsen, G. L.; Jaselkis, B.; Analyst 1992, 117, 1785.
7. Pickard, A. D.; Clarck, E. R.; Talanta 1984, 31, 763.
8. Tomcik, P.; Mrafkova, L.; Bustin. D.; Microchim. Acta 2003, 141, 69.
9. Dzyadevych, S. V.; Arkhypova, V. N.; Korpan, Y. I.; El'skaya, A. V.; Soldatkin, A. P.; Jaffrezic-Renault, N.; Martelet, C.; Anal. Chim. Acta 2001, 445, 47.
10. Korpan, Y. I.; Gonchar, M. V.; Sibirny, A. A.; Martelet, C.; El'skaya. A. V.; Gibson, T. D.; Soldatkin, A. P.; Biosens. Bioelectron. 2000, 15, 77.
11. Burini, G.; Coli, R.; Anal. Chim. Acta 2004, 511, 155.
12. Pérez-Ruiz, T.; Martínez-Lozano, C.; Tomás, V., Fenoll.; J. Anal. Bioanal. Chem. 2003, 375, 661; Pinheiro, H. L. C.; Andrade, V.; Pereira, P. A. P.; Andrade, J. B.; Microchem. J. 2004, 78, 15.
13. Reche, F.; Garrigós, M. C.; Sánchez, A.; Jiménez, A.; J. Chromatogr., A 2000, 896, 51.
14. Kato, S.; Burke, P. J.; Fenick, D. J.; Taatjes, D. J.; Bierbaum, V. M.; Koch, T. H.; Chem. Res. Toxicol. 2000, 13, 509; Kato, S.; Burke, P. J.; Koch, T. H.; Bierbaum, V. M.; Anal. Chem. 2001, 73, 2992; Rivero, R. T.; Topiwala, V.; J. Chromatogr., A 2004, 1029, 217.
15. http://www.cdc.gov/niosh/nmam/pdfs/3500.pdf, acessada em Setembro 2009.
16. Bosque-Sendra, J. M.; Pescarolo, S.; Cuadros-Rodríguez, L.; García-Campaña, A. M.; Almansa-López, E. M.; Fresenius´ J. Anal. Chem. 2001, 369, 715; Ross, P. F.; Draayer, H.; Itoh, O.; Biologicals 2002, 30, 37; Zhang, Z. Q.; Yan, H. T; Yuc, X. F.; Microchim. Acta 2004, 146, 259; Sibirny, V. A.; Gonchar, M. V.; Grabek-Lejko, D.; Pavlishko, H. M.; Csoregi, E.; Sibirny, A. A.; Int. J. Environ. Anal. Chem. 2008, 88, 289.
17. Feigl, F.; Spot tests in organic analysis, 5th ed., Elsevier: Amsterdam, 1956.
18.Georghiou, P. E.; Ho, C. K.; Can. J. Chem. 1989, 67, 871.
19. Fagnani, E.; Melios, C. B.; Pezza. L.; Pezza, H. R.; Talanta 2003, 60, 171.
20. Gigante, A. C.; Gotardo, M. A.; Tognolli, J. O.; Pezza, L.; Pezza, H. R.; Microchem. J. 2004, 77, 47.
21. Tininis, A. G.; Santini, A. O.; Fagnani, E.; Melios, C. B.; Pezza, L.; Pezza, H. R.; Eclet. Quim. 2001, 26, 175.
22. Kingston, H. M.; Jassie, L. B. ; Introduction to microwave sample preparation - theory and practice, ACS Professional Reference Books: Washington, 1988.
23. Gigante, A. C.; Pezza, L.; Pezza, H. R.; Eclet. Quim. 2002, 27, 223.
24. http://www.anvisa.gov.br/monitora/cmed/legis/comunicados/lista_conformidade.pdf , acessada em Setembro 2009.
25. United States Pharmacopeial Convention; The United States Pharmacopeia - the national formulary , 23rd ed., U.S. Pharmacopeia: Rockville, 1995.
26. Miller, J. N.; Miller, J. C.; Statistics and chemometrics for analytical chemistry, 4th ed., Pearson Education: Essex, 2000.
27. Balasubramanian, M.; Thennarassu, S.; Sudhakaran, T.; Perumal, P. T.; Biol. Pharm. Bull. 2003, 26, 1211.
28. Pavitrapok, C.; Williams, D. A.; J. Pharm. Biomed. Anal. 2006, 40, 1243.
29. OriginLab Corporation; ORIGIN®6.1; Data Analysis and Technical Graphics; Northampton, Estados Unidos, 2000.
Recebido em 6/11/08; aceito em 5/5/09; publicado na web em 30/9/09
- 1. Nickel, J. C.; J. Urol. 2005, 173, 21.
- 2. Kaszycki, P.; Koloczek, H.; Biodegradation 2002, 13, 91.
- 3. Kirk, R. E.; Othmer, D .F.; Encyclopedia of chemical technology, The Interscience Encyclopedia: New York, 1951.
- 4. Sharma, S. P.; Lahiri, S. C.; J. Indian Chem. Soc 2008, 85, 273.
- 5. Turi, E. A.; Thermal caracterization of polymeric materials, Academic Press: Orlando, 1981.
- 6. Madsen, G. L.; Jaselkis, B.; Analyst 1992, 117, 1785.
- 7. Pickard, A. D.; Clarck, E. R.; Talanta 1984, 31, 763.
- 8. Tomcik, P.; Mrafkova, L.; Bustin. D.; Microchim. Acta 2003, 141, 69.
- 9. Dzyadevych, S. V.; Arkhypova, V. N.; Korpan, Y. I.; El'skaya, A. V.; Soldatkin, A. P.; Jaffrezic-Renault, N.; Martelet, C.; Anal. Chim. Acta 2001, 445, 47.
- 10. Korpan, Y. I.; Gonchar, M. V.; Sibirny, A. A.; Martelet, C.; El'skaya. A. V.; Gibson, T. D.; Soldatkin, A. P.; Biosens. Bioelectron 2000, 15, 77.
- 11. Burini, G.; Coli, R.; Anal. Chim. Acta 2004, 511, 155.
- 12. Pérez-Ruiz, T.; Martínez-Lozano, C.; Tomás, V., Fenoll.; J. Anal. Bioanal. Chem. 2003, 375, 661;
- Pinheiro, H. L. C.; Andrade, V.; Pereira, P. A. P.; Andrade, J. B.; Microchem. J. 2004, 78, 15.
- 13. Reche, F.; Garrigós, M. C.; Sánchez, A.; Jiménez, A.; J. Chromatogr., A 2000, 896, 51.
- 14. Kato, S.; Burke, P. J.; Fenick, D. J.; Taatjes, D. J.; Bierbaum, V. M.; Koch, T. H.; Chem. Res. Toxicol 2000, 13, 509;
- Kato, S.; Burke, P. J.; Koch, T. H.; Bierbaum, V. M.; Anal. Chem. 2001, 73, 2992;
- Rivero, R. T.; Topiwala, V.; J. Chromatogr., A 2004, 1029, 217.
- 15. http://www.cdc.gov/niosh/nmam/pdfs/3500.pdf, acessada em Setembro 2009.
» link - 16. Bosque-Sendra, J. M.; Pescarolo, S.; Cuadros-Rodríguez, L.; García-Campaña, A. M.; Almansa-López, E. M.; Fresenius´ J. Anal. Chem 2001, 369, 715;
- Ross, P. F.; Draayer, H.; Itoh, O.; Biologicals 2002, 30, 37;
- Zhang, Z. Q.; Yan, H. T; Yuc, X. F.; Microchim. Acta 2004, 146, 259;
- Sibirny, V. A.; Gonchar, M. V.; Grabek-Lejko, D.; Pavlishko, H. M.; Csoregi, E.; Sibirny, A. A.; Int. J. Environ. Anal. Chem 2008, 88, 289.
- 17. Feigl, F.; Spot tests in organic analysis, 5th ed., Elsevier: Amsterdam, 1956.
- 18.Georghiou, P. E.; Ho, C. K.; Can. J. Chem 1989, 67, 871.
- 19. Fagnani, E.; Melios, C. B.; Pezza. L.; Pezza, H. R.; Talanta 2003, 60, 171.
- 20. Gigante, A. C.; Gotardo, M. A.; Tognolli, J. O.; Pezza, L.; Pezza, H. R.; Microchem. J 2004, 77, 47.
- 21. Tininis, A. G.; Santini, A. O.; Fagnani, E.; Melios, C. B.; Pezza, L.; Pezza, H. R.; Eclet. Quim. 2001, 26, 175.
- 22. Kingston, H. M.; Jassie, L. B. ; Introduction to microwave sample preparation - theory and practice, ACS Professional Reference Books: Washington, 1988.
- 23. Gigante, A. C.; Pezza, L.; Pezza, H. R.; Eclet. Quim. 2002, 27, 223.
- 24. http://www.anvisa.gov.br/monitora/cmed/legis/comunicados/lista_conformidade.pdf , acessada em Setembro 2009.
» link - 25. United States Pharmacopeial Convention; The United States Pharmacopeia - the national formulary , 23rd ed., U.S. Pharmacopeia: Rockville, 1995.
- 26. Miller, J. N.; Miller, J. C.; Statistics and chemometrics for analytical chemistry, 4th ed., Pearson Education: Essex, 2000.
- 27. Balasubramanian, M.; Thennarassu, S.; Sudhakaran, T.; Perumal, P. T.; Biol. Pharm. Bull 2003, 26, 1211.
- 28. Pavitrapok, C.; Williams, D. A.; J. Pharm. Biomed. Anal 2006, 40, 1243.
- 29. OriginLab Corporation; ORIGIN® 6.1; Data Analysis and Technical Graphics; Northampton, Estados Unidos, 2000.
Datas de Publicação
-
Publicação nesta coleção
06 Jan 2010 -
Data do Fascículo
2009
Histórico
-
Aceito
05 Maio 2009 -
Recebido
06 Nov 2008