Resumo
The use of poorly water soluble molecules in pharmaceutical area has grown. Since these molecules exhibit low oral bioavailability, they are not used in intravenous administrations. Therefore, it is necessary to develop their new formulations with the aim to increase their oral bioavailabilities as to enable intravenous applications. One of the few possibilities in achieving this is a nanonization process that can produce crystals smaller than 1 μm by high pressure homogenization and without use of organic solvents. This mini-review describes technical aspects of the nanocrystal production, morphological aspects (polymorphisms), the market relevance of the nanocrystals products that are already in clinical phase or at the market, as well as, perspectives for the near future.
drugs; homogenization; nanocrystals
drugs; homogenization; nanocrystals
REVISÃO
Tecnologia de nanocristais em fármacos
Nanocrystal technology in pharmaceuticals
Nelson Durán* * e-mail: duran@iqm.unicamp.br ; Marcela Durán; Ljubica Tasic; Priscyla D. Marcato
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas - SP, Brasil
ABSTRACT
The use of poorly water soluble molecules in pharmaceutical area has grown. Since these molecules exhibit low oral bioavailability, they are not used in intravenous administrations. Therefore, it is necessary to develop their new formulations with the aim to increase their oral bioavailabilities as to enable intravenous applications. One of the few possibilities in achieving this is a nanonization process that can produce crystals smaller than 1 μm by high pressure homogenization and without use of organic solvents. This mini-review describes technical aspects of the nanocrystal production, morphological aspects (polymorphisms), the market relevance of the nanocrystals products that are already in clinical phase or at the market, as well as, perspectives for the near future.
Keywords: drugs; homogenization; nanocrystals.
INTRODUÇÃO
Dependendo da sua solubilidade em água, cada fármaco possui um perfil diferente e único de liberação e de ação no alvo. Os fármacos pouco solúveis em água, assim como os que apresentam sérios efeitos colaterais, requerem uma tecnologia para a sua liberação em um alvo-específico. Outros problemas como lenta absorção e a aplicação de volumes excessivos em administrações intravenosas (IV) podem ser melhorados através do uso de sistemas de liberação sustentada (sistemas inteligentes).1 Isto ampliaria o uso de vários fármacos, alguns deles biotecnológicos, tais como a afidicolina (estrutura a), a violaceína (estrutura b) e o P-MAPA (estrutura c).2-5

Os fármacos pouco solúveis em sistema aquosos6 e, também, pouco solúveis em alguns meios orgânicos como, por exemplo, óleos podem ser "solubilizados" em uma emulsão óleo em água (o/a). Neste caso, o fármaco pode se localizar na interface o/a. Baseado neste conceito, Muller7 desenvolveu uma tecnologia livre de solvente capaz de incorporar fármacos na camada interfacial de emulsões óleo em água. Esta tecnologia, denominada de SolEmuls®,1 se baseia na co-homogeneização de cristais de fármacos adicionados a uma emulsão parenteral.
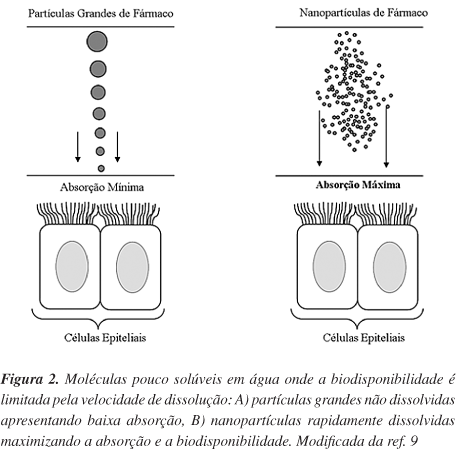
Um outro problema de fármacos pouco solúveis em meio aquoso é a sua baixa biodisponibilidade quando administrados oralmente. Este fato pode ser atribuído a dois fatores, além da sua possível degradação no aparelho digestivo: baixa velocidade de dissolução e, baixa permeabilidade do fármaco nas paredes do aparelho digestivo. Em geral, fármacos que possuem baixa solubilidade apresentam também uma baixa velocidade de dissolução. Isto pode ser explicado pela Equação de Noyes-Whitney8 que descreve a velocidade de dissolução (dc/dt) que é proporcional ao gradiente de concentração (cs-cx/h ), onde o fator cs é a concentração total do fármaco no líquido (solubilidade de saturação) e o fator h é a distância de difusão acima da superfície da partícula do fármaco. A velocidade de dissolução é dada em função da área superficial: dc/dt = DA(cs " cx)/h onde D é o coeficiente de difusão e A é a área superficial. Quanto maior a velocidade de agitação no meio de dissolução, menor será o valor de h e, por consequência, maior será a velocidade de dissolução do fármaco. A situação ideal é quando o h tende a zero e, simultaneamente, dc/dt tende para infinito (Figura 1).
Em função destas correlações, a absorção oral de fármacos pouco solúveis em água pode ser melhorada pelo aumento da velocidade de dissolução do fármaco através do aumento da sua área superficial e, aumento da solubilidade de saturação, cs, do fármaco. O primeiro aspecto pode ser atingido por micronização, entretanto para a maioria dos produtos biotecnológicos este procedimento não é suficiente devido à solubilidade muito baixa destes produtos. Desta forma, o aumento da área superficial não possibilita a obtenção de uma rápida velocidade de dissolução para atingir níveis plasmáticos terapêuticos. Porém, a transformação de microcristais do fármaco em nanocristais poderia aumentar sua absorção oral. Isto porque a redução para nanocristais, além de aumentar a velocidade de dissolução do fármaco (aumento da área superficial) (Figura 2) aumenta, simultaneamente, a sua solubilidade de saturação (cs). Deste modo, as partículas de nanocristais podem ser administradas por via oral e por via intravenosa em suspensões aquosas. Estas suspensões aquosas de nanocristais podem ser estabilizadas por tensoativos carregados (que proporcionam estabilidade eletrostática) ou não iônicos (que proporcionam estabilização estérica). A combinação de ambos tensoativos permite que dispersões de nanocristais sejam menos suscetíveis aos eletrólitos do corpo que podem provocar uma redução do potencial zeta dos cristais, favorecendo a agregação dos mesmos. Dentre os estabilizadores mais utilizados nesta aplicação e aprovados pela FDA, pode-se citar a lecitina, o Tween 80, o poloxâmero 188, o glicolato de sódio e o polivinilpirrolidona de baixa massa molar.8
Alta biodisponibilidade (até 100%) de um fármaco pode ser obtida com nanocristais com diâmetro médio entre 200-400 nm. Nesta faixa de diâmetro é possível a aplicação destes fármacos por via intravenosa, já que seu tamanho é muito menor que o diâmetro dos vasos capilares (5-6 μm). Vários métodos de nanonização são conhecidos,10 mas o mais simples é a homogeneização à alta pressão.11 Neste método, o fármaco em pó é suspenso em uma solução de tensoativos sob agitação formando uma micro/macro suspensão que é, em seguida, homogeneizada à alta pressão (DissoCubes® process).11 A nanonização também pode ser realizada pelo método denominado NanoCrystal®.9,12 Esse método se baseia na moagem de suspensões aquosas de cristais micrométricos do fármaco estabilizados por tensoativos. Outro processo que permite a formação de nanocristais em sistemas não aquosos ou em meio aquoso reduzido é a tecnologia chamada de Nanopure®. Este processo atinge a homogeneização através de um meio de dispersão à baixa pressão de vapor, onde, por exemplo, fármacos nanocristalinos podem ser obtidos em suspensão de polietilenoglicol líquido e, em seguida encapsulados em gelatina.13 Recentemente, nanosuspensões de um imunomodulador, o P-MAPA, foram obtidas através do método de homogeneização à alta pressão (Figura 3).14
Uma propriedade interessante dos nanocristais é sua estabilização devido à camada superficial nanométrica (1-2 nm) que é formada,15 por moléculas oxidadas ou hidratadas que protegem a parte interna do fármaco nanocristalino (Figura 4).
Uma outra vantagem da nanonização é o aumento da biodisponibilidade oral do fármaco nanocristalino. Esta biodisponibilidade pode ser aumentada através da incorporação do nanocristais em nanoestruturas como, por exemplo, em nanopartículas lipídicas sólidas (NLS), carregadores lipídicos nanoestruturados (CLN) ou transformado em conjugado fármaco-lipídio (CFL).1
MÉTODOS DE HOMOGENEIZAÇÃO
Os métodos mais importantes para nanonização por homogeneização serão discutidos a seguir.
Homogeneização por pistão-orifício (Dissocubes ® )
Em 1994, Müller e colaboradores11 desenvolveram um novo método para produção de nanossuspensões de fármacos utilizando como solvente a água. Este método utiliza um homogeneizador de alta pressão que contém um pistão cilíndrico que pode gerar uma pressão de até 2000 bar. A suspensão é pressionada através de um orifício de 3-5 μm a uma pressão entre 150-1500 bar. Segundo a Equação de Bernoulli,16 no orifício da homogeneização a pressão dinâmica (velocidade de fluxo) aumenta e, simultaneamente, diminui a pressão estática sobre o líquido. Nestas condições o líquido começa a ferver formando bolhas de gás que explodem quando a suspensão deixa o orifício. Em seguida, a pressão volta ao normal (cavitação). A formação de bolhas de gás seguida por suas explosões gera ondas de choque que causam a diminuição das partículas (Figura 5).
A homogeneização por pistão-orifício pode ser utilizada para a produção de nanossuspensões; entre os equipamentos disponíveis no mercado os mais utilizados são APV Gaulin, Avestin e Niro Soavi. Esta mesma tecnologia de homogeneização à alta pressão foi adquirida pela Skypharma PLC no final dos anos 90 e aplicada no desenvolvimento de suas formulações.
Tecnologia Nanopure ®
Em alguns casos, a administração oral de nanocristais é mais conveniente em meio não aquoso como em óleo, em cápsulas de gelatina ou em grânulos.17 Entretanto, a preparação de formulações semi-sólidas/sólidas a partir de suspensões aquosas de nanocristais requer, em geral, a remoção da água por evaporação. Porém, é possível a obtenção direta destas formulações por homogeneização em um meio não-aquoso (Nanopure®). A tecnologia de homogeneização não aquosa ou Nanopure® foi estabelecida contra a ideia de que a cavitação é a principal força responsável pela diminuição do tamanho dos cristais na homogeneização à alta pressão. Além disto, através da técnica de Nanopure® é possível ter a mesma eficiência de redução dos cristais em relação aos métodos aquosos. Utilizando esta técnica, fármacos nanocristalinos dispersos em polietilenoglicol líquido ou em óleos podem ser comercializados como produtos encapsulados em cápsulas de gelatina ou de hidroxipropilmetil celulose (HPMC).17 Além disto, é possível a obtenção de nanossuspensões isotônicas prontas para administração intravenosa através da homogeneização do fármaco em uma mistura de água e glicerol.
Tecnologia SolEmuls ®
Na tecnologia SolEmuls® (solubilização por emulsificação) o fármaco é disperso em uma emulsão óleo em água (o/a) contendo o estabilizante lecitina. Em seguida, esta mistura é homogeneizada à alta pressão obtendo-se uma formulação intravenosa de nanocristais. O fármaco pouco solúvel fica na interface o/a.1,17 Esta tecnologia já foi utilizada no preparado de formulações intravenosas de diversos fármacos como, por exemplo, a anfotericina B (estrutura d), itraconazol (estrutura e), cetoconazol (estrutura f), carbamazepina18 (estrutura g), cetoprofeno19 (estrutura h), nifedipina20 (estrutura i), ucb-35440-321 (estrutura j), hidrocortisona (estrutura k), prednisolona (estrutura l) ou dexametasona22 (estrutura m), acetato de hidrocortisona23,24 (estrutura n). A obtenção de nanoemulsões com a maioria destes compostos é complicada devido à baixa solubilidade dos mesmos. Entretanto, este problema foi resolvido pela tecnologia SolEmuls®; utilizando-se uma simples co-homogeneização, como no caso da anfotericina B. Este composto em pó foi inicialmente adicionado, sob agitação, em uma pré-emulsão óleo em água estabilizada por lecitina. Em seguida, esta pré-emulsão foi homogeneizada a 600 bar. A alta velocidade de fluxo no processo de homogeneização resultou em uma rápida dissolução dos nanocristais e alterou a partição do fármaco na interface o/a. O número de ciclos de homogeneização depende do diâmetro inicial dos cristais sendo que, em geral, de 5-20 ciclos são necessários. Entretanto, o número de ciclos de homogeneização pode ser reduzido se os cristais forem previamente reduzidos a diâmetros menores. A redução do diâmetro dos cristais pode ser realizado por moinho de bolas.17
Na preparação de nanocristais pela tecnologia de SolEmuls® é utilizado ao redor de 20-30% do fármaco nanocristalino numa forma de nanoemulsão. As nanossuspensões podem ser estabilizadas por lecitina por outros tensoativos, tais como Tween 80 ou o Poloxamero 188.18

Tecnologia NanoCrystal ®
A tecnologia de NanoCrystal® tem sido utilizada na preparação de nanocristais. Este método consiste no preparo de uma dispersão do fármaco com um estabilizador, sendo a água o solvente mais utilizado. Em seguida, esta dispersão é processada por moagem de alta energia de moagem com poliestireno altamente ramificado, que proporciona um meio de prolongada moagem, resultando na eficiente formação de cristais nanométricos. Este processo, aprovado em vários países, é versátil, pode ser aplicado em diferentes compostos aumentando sua solubilidade em água e permitindo a obtenção de formulações de nanocristais para administrações oral e intravenosa.9 Através deste processo, nanocristais de diferentes fármacos foram preparados como o cilostazol (estrutura o), que apresentou um aumento na sua biodisponibilidade quando aplicado em cachorros Beagle.25 Outros exemplos incluem iboprufeno26 (estrutura p), budesonida27 (estrutura q), espironolactona28 (estrutura r), insulina29 (estrutura s), itraconazol (estrutura e), hidroxi-itraconazol30 (estrutura t) e Mk-086931 (estrutura u).


POLIMORFISMO NOS NANOCRISTAIS
Um aspecto importante no estudo de nanonização dos fármacos é a forma dos nanocristais formados, visto que a estrutura cristalina (forma) destes pode modificar vários aspectos da sua ação, assim como suas estabilidade e biodisponibilidade.32-37 O polimorfismo (existência de diferentes formas cristalinas de um mesmo fármaco) pode causar variações no ponto de fusão, estabilidade, densidade e na solubilidade do fármaco, já que estas propriedades dependem da tendência de escape da molécula de uma estrutura cristalina particular. Além disto, a forma polimórfica influencia na concentração de saturação (cs) sendo que a solubilidade é maior para as modificações polimórficas que apresentam alta energia interna e baixo ponto de fusão.38
Em geral, sabe-se que a forma mais cristalina de um polimorfo é a mais estável devido, principalmente, a sua baixa energia livre, o seu alto ponto de fusão e a sua baixa solubilidade.33
No caso do fármaco nifedipina (estrutura i), foram identificadas três formas polimórficas através da análise térmica. A forma polimórfica I apresenta um ponto de fusão de 172 ºC, a forma II de 163 ºC e a forma III de 140 ºC. A forma polimórfica I é a mais estável e é caracterizada pela menor solubilidade em água. Através da técnica de calorimetria diferencial de varredura (DSC) foi observado que a forma comercial não moída e os nanocristais deste fármaco estão presentes na forma polimórfica I. Além disto, foi verificado, por difração de raios-X, que o processo de homogeneização não alterou o estado cristalino do fármaco, já que o perfil de difração deste fármaco foi conservado após a homogeneização.20 Outra técnica que pode ser utilizada na caracterização do polimorfismo é a microscopia de força atômica (AFM), que permite a visualização da morfologia dos cristais.34,36
Outros aspectos relevantes na obtenção de nanocristais são: a formação de uma fração amorfa no cristal ou cristais completamente amorfos. Isto é comumente obtido quando os cristais são reduzidos utilizando-se o processo de homogeneização à alta pressão. Por exemplo, no processo de formação de nanocristais do fármaco RMKP22 (estrutura ii) pela técnica de NanoCrystal® foi observado a formação de cristais no estado amorfo. No entanto, quando foi utilizado o processo de moagem de baixa energia foram observadas estruturas completamente cristalinas. A extensão da fração amorfa produzida através da homogeneização à alta pressão depende dos parâmetros de produção como, por exemplo, a pressão de homogeneização, o número de ciclos e a dureza do fármaco. O aumento da fração amorfa leva a um aumento da concentração de saturação, já que os fármacos amorfos apresentam uma solubilidade de saturação maior que as formas cristalinas.37 Logo, o aspecto de polimorfismo é importante e deve ser monitorado nas nanoformulações, devido às implicações tanto na solubilidade como nos outros parâmetros farmacocinéticos.37

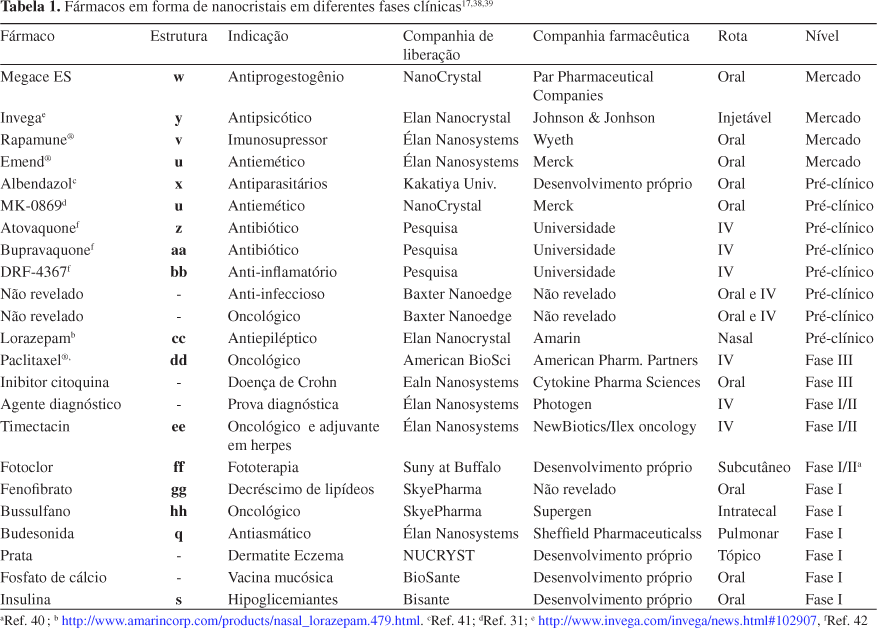
PRODUTOS NO MERCADO E EM FASE CLÍNICA
No caso dos nanocristais o período entre a pesquisa (invenção) e o primeiro produto no mercado foi extremamente curto, quando comparado ao desenvolvimento de outros tipos de fármacos. Por exemplo, os lipossomas foram inventados em 1912 por Lehmann e reinventados em 1968 por Bangham, porém os primeiros produtos só apareceram no mercado nos anos 90, ou seja, após 20 anos. As primeiras patentes de nanocristais foram registradas nos anos 90 pela companhia Nanosystems (hoje Élan). O primeiro produto Rapamune® foi disponibilizado no mercado em 2000 pela companhia Wyeth na forma de comprimido (contendo 1 ou 2 mg de Sirolimus) (estrutura v) ou na forma de solução. A forma sólida do Rapamune® apresenta 21% mais biodisponibilidade do que a forma em solução devido, exclusivamente, aos nanocristais presentes na sua formulação. O segundo produto lançado no mercado foi o Emend® (estrutura u). Este foi introduzido em 2001 pela companhia Merck na forma cápsulas (125 mg do fármaco Aprepitant17) (estrutura u).
Por muito tempo esta tecnologia não foi, praticamente, utilizada pela indústria devido ao escalonamento da produção (produção em larga escala). A situação mudou com avanços da tecnologia de nanonização que introduziu mais facilidades neste tipo de produção. Isto levou a uma aceleração do desenvolvimento de formulações baseadas em nanocristais devido, principalmente, à concorrência e à dinâmica do mercado de fármacos e cosméticos por novas tecnologias (Tabela 1).
Dentre as empresas que utilizam a tecnologia de nanonização pode-se citar a Élan que produz nanossuspensões aplicando moinhos de pérolas, a companhia Baxter que estabeleceu uma linha de produção asséptica baseada em um homogeneizador de pistão-orifício para a plataforma de produtos da linha Nanoedge®.
Esta área tem atingido uma grande importância no crescimento de novas indústrias, como pode ser observado na Tabela 2.
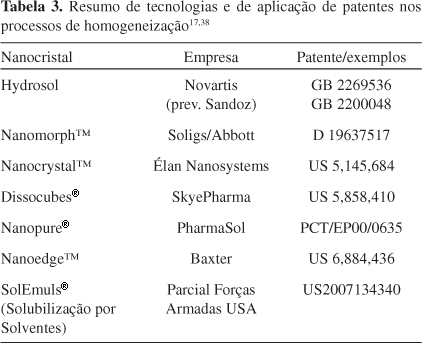
Tabela 3 mostra o resumo das diferentes tecnologias e patentes de empresas, como Novartis, Aolig/Abbott, Élan, Nanosystems, SkyePharma, PharmaSol, Baxter, que aplicaram o processo de homogeneização à alta pressão na obtenção de novas formulações de fármacos já conhecidos.
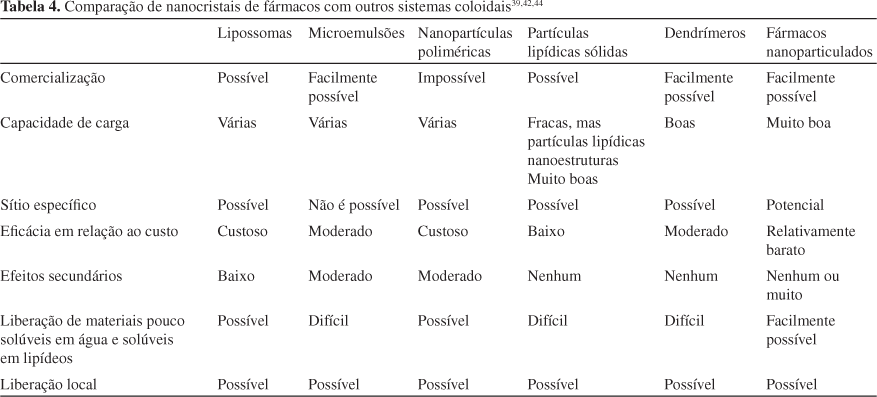
Apesar do grande desenvolvimento de sistemas nanoparticulados, sua comercialização ainda apresenta vários problemas como, por exemplo, escalonamento e os custos da produção, além dos requerimentos exigidos pelos órgãos regulatórios. Por exemplo, poucas partículas poliméricas foram comercializadas 30 anos após seu descobrimento. Este fato, provavelmente, se deve à ênfase que a indústria farmacêutica deu à produção de nanocristais de fármacos, em vez de sistemas de encapsulamento nanoparticulados. Tudo isto leva a pensar que provavelmente a razão principal para este paradigma seja o surgimento de tecnologias adequadas em escala industrial; entretanto, é evidente que os fármacos nanoparticulados oferecem várias vantagens sobre outros sistemas coloidais. A Tabela 4 resume a comparação de nanocristais de fármacos com os sistemas carreadores de fármacos.39,42,44
CONCLUSÃO E PERSPECTIVAS
Um dos empecilhos mais importantes no desenvolvimento de nanofármacos é o cumprimento das normas regulatórias. Entretanto, nos últimos anos uma grande atenção tem sido dada aos fármacos hidrofóbicos, já que as normas da FDA são cumpridas mais facilmente em comparação com os sistemas carreadores nanoparticulados (Tabela 1). O processo que evidentemente teve um grande impacto no desenvolvimento de novos fármacos foi o de moagem e a homogeneização à alta pressão. É importante mencionar que as tecnologias descritas na Tabela 2 representam uma segunda geração de nanonização de fármacos hidrofóbicos. Na opinião de Date e Patravale,42 entre todas as metodologias mencionadas, a transferência de massa por antissolvente supercrítico e a precipitação reativa de alta gravidade são tecnologias com grande potencial de uso no futuro, devido a sua simplicidade, fácil escalonamento e eficiência no processo de nanonização. Entretanto, a história mostrou que estes métodos não foram desenvolvidos na mesma intensidade como os de homogeneização à alta pressão.
Das e Das33 apontam que a solubilização de fármacos pouco solúveis em sistemas aquosos continua colocando desafios à indústria farmacêutica, entretanto, as diversas tecnologias que agora estão disponíveis para a reformulação destes terapêuticos permitem a sua eficiente aplicação. Evidentemente, esta estratégia pode ser custosa e proibitiva para pequenas empresas farmacêuticas.
Por outro lado, Keck e Müller,17 afirmam que independentemente da maneira da produção, os fármacos nanocristalinos têm grandes vantagens como, por exemplo, a utilização de sistemas simples de produção. Isto é de suma importância já que quanto mais complicado é o sistema de produção, mais tempo este produto ficará longe do mercado e dos pacientes. Sistemas simples podem chegar, rapidamente, a ser uma realidade, como foi mostrado pelo primeiro produto nanocristalino no mercado.
Segundo Merisko-Liversidge e Liversidge,9 a tecnologia nas formulações com nanocristais deixou novas estratégias para resolver aspectos associados com moléculas pouco solúveis em água. A homogeneização permitiu avançar rapidamente em estudos de medidas de eficácia e de segurança de novos fármacos. Além disto, esta tecnologia possibilitou o desenvolvimento de novos produtos através de processos escalonáveis e com grande possibilidade de atingir o mercado. Isto obviamente pode permitir estudos com poucas doses, porém, com máxima exposição do fármaco no paciente. Estratégias de formulação de fármacos nanoparticulados são necessidade eminente na indústria farmacêutica. Isto permitiria abrir novos caminhos ou alternativas para abordar necessidades médicas ainda não satisfeitas. É provável que estas estratégias que são importantes agora como, por exemplo, seleção de fármacos devido a sua solubilidade, mude para aspectos mais relacionados com a eficácia e segurança dos mesmos. Isto talvez diminua o tempo de desenvolvimento e possibilite o surgimento de novas terapias e agentes de diagnóstico para doenças mais desafiadoras que tenham que ser controladas ou erradicadas. Como é evidente que a era da nanotecnologia na indústria farmacêutica já começou, durante as próximas décadas será interessante verificar se todas as promessas previstas chegaram a ser realidades.

Nossa experiência em homogeneização à alta pressão na produção de nanocristais permite dizer que o método tem grande versatilidade, permitindo trabalhar tanto com fármacos hidrossolúveis quanto com lipossolúveis, e com diversos solventes, desde água a solventes orgânicos, assim como com diversos tensoativos. Além disto, este processo também permite a produção de nanocristais em larga escala e estéreis que são aspectos importantes em aplicações na indústria farmacêutica e cosmética.
AGRADECIMENTOS
Agradecemos o apoio da FAPESP, a Rede de Nanocosméticos (MCT/CNPq) e a Farmabrasilis (Dr. I. Nunes).
Berlin.
Recebido em 22/10/08; aceito em 28/5/09; publicado na web em 8/12/09
- 1. Shah, P.; MRS Bull. 2006, 31, 894.
- 2. Muller, R. H.; Keck, C. M.; J. Biotechnol. 2004, 113, 151.
- 3. Kayser, O.; Kiderlen, A. F.; Folkens, U.; Kolodziej, H.; Planta Med.1999, 65, 316.
- 4. Durán, N.; Justo, G. Z.; Ferreira, C. V.; Melo, P. S.; Cordi, L.; Martins, D.; Biochem. Appl. Biotechnol 2007, 48, 127.
- 5. Durán, N.; Justo, G. Z.; Nunes, I.; Patente Brasil , PIBr 2004, 0.305.373-3.
- 6. Horn, D.; Rieger, J.; Angew. Chem., Int. Ed 2001, 40, 4330.
- 7. Muller, R. H.; Patent, 2001, PCT/EPO/08726 PharmaSol GmbH
- 8. Müller, R. H.; Schmidt, S.; Buttle, I.; Akkar, A.; Schmitt, J.; Brömer, S.; Int. J. Pharm. 2004, 269, 293.
- 9. Merisko-Liversidge, E.; Liversidge, G. G.; Toxicol. Pathol. 2008, 36, 43.
- 10. Jia, L.; Wong, H.; Cesario Cerna, C.; Weitman, S. D.; Pharm. Res. 2002, 19, 1091.
- 11. Muller, R. H.; Becker, R.; Knuss, B.; Peters, K.; US Patent, 1999, 5,858,410.
- 12. Merisko-Liversidge, E.; McGurk, S.; Liversidge, G. G.; Pharm. Res. 2004, 21, 1545.
- 13. Muller, R. H.; Krause, K.; Mader, K.; Patent; 2000, WO 01/03670 A1.
- 14. Durán, N.; Marcato, P. D.; Durán, M.; Patente Brasil PIBr 2008, submetida.
- 15. Kipp, J. E.; Int. J. Pharm. 2004, 284, 109.
- 16. Muller, R. H.; Jacobs, C.; Kayser, O. Em Pharmaceutical Emulsions and suspensions; Nielloud, F.; Marti-Mestres, G., eds; Marcel Decker Press: Basel, 2000, p. 383.
- 17. Keck, C. M.; Muller, R. H.; Eur. J. Pharm. Biopharm 2006, 62, 3.
- 18. Muller, R. H.; Junghanns, J-U. A. H. Em Nanoparticulates as Drug Carriers; Torchilin V. P., ed.; Imperial College Press: Covent Garden, 2006, p. 307.
- 19. Vergote, G. J.; Vervaet, C.; Van Driessche, I.; Hoste, S.; De Smedt, S.; Demeester, J.; Jain, R. A.; Ruddy, S.; Remon, J. P.; Int. J. Pharm. 2002, 240, 79.
- 20. Hecq, J.; Deleers, M.; Fanara, D.; Vranckx, H.; Amighi, K.; Int. J. Pharm 2005, 299, 167.
- 21. Hecq, J. ; Deleers, M.; Fanara, D.; Vranckx, H. ; Boulanger, P. ; Le Lamer, S.; Amighi, K.; Eur. J. Pharm. Biopharm 2006, 64, 360.
- 22. Kassem, M. A.; Abdel Rahmanb, A. A.; Ghorab, M. M.; Ahmeda, M. B.; Khalil, R. M.; Int. J. Pharm. 2007, 340, 126.
- 23. Moschwitzer, J.; Muller, R. H.; Eur. J. Pharm. Biopharm. 2006, 62, 282.
- 24. Moschwitzer, J.; Muller, R. H.; J. Nanosci. Nanotechnol. 2006, 6, 3145.
- 25. Jinno, J. I.; Kamada, N.; Miyake, M.; Yamada, K.; Tadashi, M. T.; Odomi, M.; Toguchi, H.; Liversidge, G. G.; Higaki, K.; Kimura, T.; J. Control. Release 2006, 111, 56.
- 26. Kocbek, P.; Baumgartner, S.; Kristl, J.; Int. J. Pharm. 2006, 312, 179.
- 27. Kraft, W. K.; Steiger, B.; Beussink, D.; Quiring, J. N.; Fitzgerald, N.; Greenberg, M. D.; Waldman, S. A.; J. Clin. Pharmacol. 2004, 44, 67.
- 28. Langguth, P.; Hanafy, A.; Frenzel, D.; Grenier, P.; Nhamias, A.; Ohlig, T.; Vergnault, G.; Span-Langguth, H.; Drug Dev. Ind. Pharm. 2005, 31, 319.
- 29. Merisko-Liversidge, E.; Liversidge, G. G.; Cooper, E. R.; Eur. J. Pharm. Sci. 2003, 18, 113.
- 30. Mouton, J. W.; van Peer, A.; de Beule, K.; Vliet, V.; Donnelly, J. P.; Soons, P. A.; Antimicrob. Agents Chemother. 2006, 50, 1096.
- 31. Wu, Y.; Loper, A.; Landis, E.; Hettrick, L.; Novak, L.; Lynn, K.; Chen, C.; Thompson, K.; Higgins, R.; Batra, U.; Shelukar, S.; Kwei, G.; Storey, D.; Int. J. Pharm. 2004, 285, 135.
- 32. Fantin, G.; Fogagnolo, M.; Bortolini, O.; Masciocchi, N.; Galli, S.; Sironi, A.; New J. Chem 2003, 27, 1794.
- 33. Das, N. G.; Das, S. K.; Drug Deliv. Rep., 2006, Spring/Summer, 52.
- 34. Shi, H. G.; Farber, L.; Michaels, J. M.; Dickey, A.; Thompson, K. C.; Shelukar, S. D.; Hurter, P. N.; Reynolds, S. D.; Kaufman, M. J.; Pharm. Res. 2003, 20, 479.
- 35. Danesh, A.; Chen, X.; Davies, M. C.; Roberts, C. J.; Sanders, G. H. W.; Tendlers, S. J. B.; Williams, P. M.; Langmuir 2000, 16, 866.
- 36. Danesh, A.; Chen, X.; Davies, M. C.; Roberts, C. J.; Sanders, G. H. W.; Tendlers, S. J. B.; Williams, P. M.; Wilkins, M. J.; Pharm. Res. 2006, 17, 887.
- 37. Muller, R. H.; Jacobs, C.; Kayser, O.; Advan. Drug Deliv. Rev .2001, 47, 3.
- 38. Rabinow, B. E.; Nat. Rev. Drug Discov 2004, 3, 785.
- 39. Marcato, P. D.; Durán, N.; J. Nanosci. Nanotechnol. 2008, 8, 2016.
- 40. Baba, K.; Pudavar, H. E.; Roy, I.; Ohulchanskyy, T. Y.; Chen, Y.; Pandey, R. K.; Prasad, P.N.; Mol. Pharm. 2007, 4, 289.
- 41. Kumar, M. P.; Rao, Y. M.; Apte, S.; Curr. Nanosci. 2008, 4, 53.
- 42. Date, A. A.; Patravale, V. B.; Curr. Opin. Colloid Interface Sci 2004, 9, 222.
- 43. Baba, K.; Kasai, H.; Okada, S.; Oikawa, H.; Nakanishi, H.; Opt. Mater. 2003, 2, 591.
- 44. Marcato, P. D.; Rev. Eletrônica Farmácia 2009, 6,1.
Datas de Publicação
-
Publicação nesta coleção
05 Fev 2010 -
Data do Fascículo
2010
Histórico
-
Aceito
28 Maio 2009 -
Recebido
22 Out 2008