Resumo
An UV-Vis spectrophotometry analytical method for quantifying Efavirenz was developed and validated as an alternative to replace the HPLC current method. The report method presents sample concentration of 10 μg mL-1, dissolved in a solution ethanol:water (60:40, v/v), economic and technically adequate for the purpose adopted. The results and the statistical treated proved that the method being considered an precise and accurate analytical low cost alternative for laboratory routine. The adaptability of this method in product and other analytical methods development has been challenged by mathematical calculation of drug extinction coefficient in water and methanol and practical experiments, showing interesting results.
UV-Vis spectroscopy; Efavirenz; analytical method
UV-Vis spectroscopy; Efavirenz; analytical method
NOTA TÉCNICA
Desenvolvimento de método analítico para quantificação do efavirenz por espectrofotometria no UV-Vis
UV-Vis spectrophotometry analytical method development for quantifying efavirenz
Lariza Darlene Santos AlvesI; Larissa Araújo RolimI; Danilo Augusto Ferreira FontesI; Pedro José Rolim-NetoI, * * e-mail: pedro.rolim@pq.cnpq.br ; Mônica Felts de La Roca SoaresII; José Lamartine Soares SobrinhoII
IDepartamento de Ciências Farmacêuticas, Universidade Federal de Pernambuco, Rua Arthur de Sá, s/n, 50740-521 Recife - PE, Brasil
IIDepartamento de Bioquímica e Farmacologia, Universidade Federal do Piauí, Campus Universitário Ministro Petrônio Portella, 64049-550 Teresina - PI, Brasil
ABSTRACT
An UV-Vis spectrophotometry analytical method for quantifying Efavirenz was developed and validated as an alternative to replace the HPLC current method. The report method presents sample concentration of 10 μg mL-1, dissolved in a solution ethanol:water (60:40, v/v), economic and technically adequate for the purpose adopted. The results and the statistical treated proved that the method being considered an precise and accurate analytical low cost alternative for laboratory routine. The adaptability of this method in product and other analytical methods development has been challenged by mathematical calculation of drug extinction coefficient in water and methanol and practical experiments, showing interesting results.
Keywords: UV-Vis spectroscopy; Efavirenz; analytical method.
INTRODUÇÃO
O Efavirenz (EFZ) é um fármaco inibidor da transcriptase reversa não nucleosídico, utilizado desde 1998 em associação com outros agentes antirretrovirais no tratamento da infecção pelo vírus da imunodeficiência humana.1 Atualmente é utilizado por cerca de 75 mil das 200 mil pessoas em terapia antirretroviral no Brasil, chegando a representar um custo de US$ 42.930.000 em 2007. Contudo, o licenciamento compulsório, ocorrido no final deste mesmo ano, possibilitou ao Brasil a produção nacional do medicamento, garantindo o acesso universal a um número cada vez maior de pacientes, reduzindo custos e viabilizando a sustentabilidade do Programa de Doenças Sexualmente Transmissíveis.2
No Brasil, assim como em grande parte do mundo, este fármaco é considerado como de primeira escolha, associado aos fármacos zidovudina e lamivudina na terapia antirretroviral; exceto para gestantes e crianças menores de 3 anos. Essa opção está fundamentada na elevada potência da supressão viral, comprovada eficácia a longo prazo e ao menor risco de efeitos adversos.2
Na literatura científica internacional, diversos métodos analíticos para a quantificação do EFZ são preconizados utilizando-se cromatografia líquida de alta eficiência (CLAE) do fármaco isolado e na forma farmacêutica cápsula,3 assim como em plasma na forma isolada e associado a outros antirretrovirais.4-14 Contudo, nenhum método analítico por espectrofotometria de absorção no ultravioleta visível (UV-vis) foi relatado.
Esta técnica é reconhecida pelas vantagens relacionadas ao seu uso, sendo utilizada principalmente no controle de qualidade na indústria farmacêutica, que exige rapidez e confiabilidade nos resultados.15 Além disso, possui baixo custo operacional, sendo de fácil utilização e produz resultados de interpretação bastante simples.16
Paralelamente, a necessidade de proporcionar qualidade às medições químicas está sendo cada vez mais reconhecida e exigida.15 Logo, para garantir que um novo método analítico gere informações confiáveis, o mesmo deve passar por uma avaliação denominada validação.
A validação é um processo contínuo que começa no planejamento da estratégia analítica e continua ao longo de todo o desenvolvimento e transferência de um método. Para registro de novos produtos, o órgão regulador do Brasil, a Agência Nacional de Vigilância Sanitária (ANVISA), e o dos Estados Unidos da América, o Food and Drug Administration (FDA), exigem a validação das metodologias analíticas e, para isso, são estabelecidos documentos oficiais que direcionam os critérios a serem adotados nesse processo.17
Logo, a validação tornou-se uma ferramenta de qualidade e não mais um proforme inflexível e automatizado. Com o desenvolvimento do setor de Desenvolvimento Analítico dentro do Controle de Qualidade, os técnicos ganharam flexibilidade para adaptar os métodos antigos, investigar desvios da qualidade e/ou desenvolver novos métodos para os fármacos de interesse.
Na atualidade, o fundamento da técnica analítica e a aplicação de equações conhecidas acelera a investigação e otimização dos métodos e práticas aplicadas. Aplicando-se o cálculo de absortividade molar ao método desenvolvido e validado para o fármaco EFZ pode-se relativizar qualquer resultado gerado em absorvância para qualquer sistema de solvente empregado, gerando agilidade e flexibilidade ao setor de Desenvolvimento Analítico.
Portanto, esse estudo descreve o desenvolvimento e a validação de um método analítico para quantificação da matéria prima EFZ, de forma que o método atenda às exigências da International Conference on Harmonization (ICH Q2A e ICH Q2B)18,19 e da ANVISA (RE n° 899 de 2003),20 conferindo praticidade, confiabilidade e baixo custo para a utilização deste método na rotina laboratorial da indústria farmacêutica.
PARTE EXPERIMENTAL
Material e métodos
Matéria-prima, reagentes e vidrarias
Para as etapas correspondentes ao desenvolvimento do método e sua validação, foram utilizadas a matéria-prima EFZ (Cristália®, lote: 1289/07, teor 98.00%) e o seu respectivo padrão de trabalho (Xiamem Mchem®, lote 050501, teor 98.32%) e a matéria-prima zidovudina (Northeast® lote: DY 070041, teor 97.88%) para a determinação da especificidade do método desenvolvido. Os reagentes utilizados foram acetonitrila (Vetec®, lote 0806979); metanol (Cinética®, lote 11850); álcool etílico absoluto (Dinâmica®, lote 30065 e Vetec®, lote 0806654); ácido clorídrico (Dinâmica®, lote 10023) e água ultrapurificada obtida por Milli-Q Millipore® (Milli-Q® System, Massachusetts, USA). Utilizaram-se vidrarias volumétricas calibradas com certificado de calibração por lote do fabricante Satelit®.
Equipamentos
Os equipamentos utilizados foram balança analítica Bioprecisa®, modelo FA2104N; espectrofotômetro UV-Vis B582 Micronal®; agitador magnético SBSR e ultrassom 1.500 Barnson® (70 W de potência). Para a reprodutibilidade foram utilizados os seguintes equipamentos: balança analítica CT 225 Sartorius®; espectrofotômetro UV-Vis modelo 50 Vankel® e ultrassom 2.510 Barnson®.
Desenvolvimento do método analítico
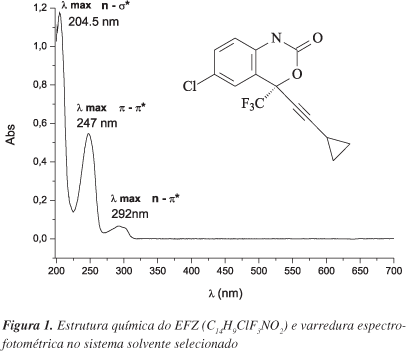
Primeiramente realizou-se um teste de solubilidade (a 25 °C) com diferentes solventes (água, álcool etílico absoluto, metanol, acetonitrila e ácido clorídrico 0,1 M), de acordo com a Farmacopeia Brasileira.21 Neste estudo observou-se o comportamento do fármaco em diferentes solventes, com objetivo de definir a melhor solução diluente para o método analítico desenvolvido. Para tal, levou-se em consideração o poder de solubilização do fármaco, custo e toxicidade do solvente e a sensibilidade do fármaco frente ao comprimento de onda utilizado. Diante disto, realizou-se uma varredura na faixa de 200 a 700 nm nos diferentes sistemas de solventes propostos (Tabela 1), para a identificação do comprimento de onda que apresentaria o valor de absorbância mais adequado para o método. A calibração do equipamento espectrofotômetro foi realizada através de padrões de referência rastreados pelo National Institute of Standards and Technology (NIST).
Posteriormente foram realizadas comparações entre as amostras obtidas pelas técnicas de agitação por ultrassonicação e agitação magnética (ambas sem aquecimento), paralelamente à variação do tempo de agitação (5, 10 e 15 min para ultrassonicação e 10 e 15 min para agitação magnética). Além disso, realizou-se a verificação da estabilidade das amostras, no sistema de solvente escolhido, avaliando-se o aspecto visual e a absorbância das mesmas, preparadas em triplicata no intervalo de 0, 24 e 48 h, na presença e ausência de luz.
Preparação da solução amostra
As amostras de EFZ foram analiticamente pesadas e solubilizadas em álcool etílico absoluto, ultrassonicadas por 10 min, obtendo-se uma concentração final de 500 μg mL-1, após aferição do balão volumétrico. Uma alíquota foi retirada dessa solução para diluição, utilizando-se como sistema de solventes álcool etílico absoluto:água purificada (60:40, v/v), obtendo-se uma concentração final de 10 μg mL-1. As amostras foram preparadas em triplicata e lidas no comprimento de onda de 247 nm.
Preparação da curva controle
Partindo da solução estoque do padrão de EFZ (500 μg mL-1), foram realizadas diluições com o sistema álcool etílico absoluto:água purificada (60:40, v/v) para a obtenção das seguintes concentrações: 8, 10 e 12 μg mL-1. A curva controle foi preparada diariamente e utilizada para os cálculos das concentrações.
Validação do método analítico
Parâmetros avaliados
No processo de validação desta metodologia analítica foram avaliados os seguintes parâmetros: robustez, linearidade, intervalo, limite de detecção, limite de quantificação, precisão, exatidão e especificidade, de acordo com as normas estabelecidas pelo ICH18,19 e pela RE n° 899/03 (ANVISA).20
A confiabilidade dos parâmetros avaliados pode ser observada pelo coeficiente de variação (CV%) ou desvio padrão relativo de uma série de medidas. Para cada parâmetro avaliado, foi determinado um CV% menor que 5%20 e tratados estatisticamente por Análise de Variância (ANOVA) One-Way e teste t de Student com um nível de significância de 95%.
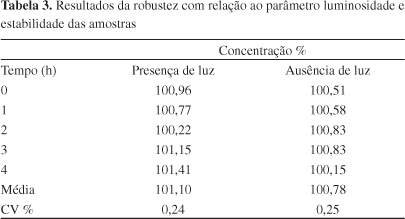
O ensaio para determinação da robustez foi realizado a partir da variação dos seguintes parâmetros: fabricante do álcool etílico absoluto (Dinâmica® e Vetec®), influência da luminosidade (presença e ausência de luz) e tempo de preparação das amostras (0, 1, 2, 3, 4 h).
A linearidade do método foi verificada a partir da análise de três curvas autênticas nas concentrações de 5, 8, 9, 10, 11, 12 e 15 μg mL-1 de EFZ. Os resultados obtidos foram tratados estatisticamente através do cálculo de regressão linear pelo método dos mínimos quadrados. Também foram estimados os limites de detecção (LD) e quantificação (LQ) de acordo com as equações LD = DP x 3/IC e LQ = DP x 10/IC, onde DP é o desvio padrão do intercepto com o eixo do Y obtido com as três curvas de linearidade e IC é a média dos coeficientes angulares das respectivas curvas. Para calculá-los, foi considerado o desvio padrão da reta com relação à absorbância do primeiro nível de concentração de EFZ das três curvas de calibração. Apesar desses parâmetros não serem considerados como requisitos necessários para testes da categoria I de validação (Testes quantitativos para a determinação do princípio ativo em produtos farmacêuticos e matérias-primas),20 os mesmos foram realizados para a utilização em uma posterior validação de limpeza de equipamentos.
A precisão do método foi avaliada nos níveis de repetitividade (precisão intracorrida), precisão intermediária (precisão intercorridas) e reprodutibilidade (precisão interlaboratorial). A repetitividade foi verificada por 6 determinações individuais a 100% da concentração teste (10 μg mL-1), analisadas em um pequeno intervalo de tempo. Para o parâmetro precisão intermediária, 3 réplicas foram analisadas variando-se os dias e os analistas, ambas também na concentração de 100%. A reprodutibilidade foi realizada em dois laboratórios diferentes em triplicata de amostras.
A exatidão foi definida através da manipulação de amostras preparadas em triplicata nas concentrações de 80, 100 e 120% da concentração teórica do EFZ, sendo determinada após o estabelecimento da linearidade e intervalo do método.
O parâmetro especificidade foi verificado através da contaminação da solução mãe de 500 μg mL-1 de EFZ com 1 mg do antirretroviral zidovudina, com o objetivo de observar a especificidade do método frente a uma possível contaminação, já que ambos são manipulados na mesma área de produção de uma indústria. A solução mãe contaminada foi diluída para se obter uma solução na concentração de 10 μg mL-1. Também foi preparada uma solução amostra não contaminada, contendo apenas o EFZ na mesma concentração e outra solução numa concentração de 0,2 μg mL-1 de zidovudina, correspondente à concentração final deste fármaco na solução contaminada de EFZ após diluição.
Determinação da absortividade molar
A espectrofotometria é fundamentada na lei de Lambert-Beer, que é a base matemática para medidas de absorção de radiação por amostras no estado sólido, líquido ou gasoso, nas regiões ultravioleta, visível e infravermelho do espectro eletromagnético. A absortividade molar (ε) é uma grandeza característica da espécie absorvente, cuja magnitude depende do comprimento de onda da radiação incidente.21 As amostras para a determinação da absortividade molar do EFZ nos solventes água purificada e metanol foram preparadas utilizando-se a mesma técnica descrita anteriormente, variando-se apenas o tempo de sonicação das amostras preparadas em água (4 h). Para as leituras das amostras, também se utilizou o comprimento de onda de 247 nm do método desenvolvido e validado.
Verificação da aplicabilidade da absortividade molar
Por meio dos três parâmetros utilizados na covalidação do método analítico, verificou-se estatisticamente a diferença dos resultados teóricos obtidos por meio do cálculo da absortividade molar e dos resultados práticos realizados com os dois solventes, água purificada e metanol. Tais parâmetros foram executados de forma idêntica à validação do método desenvolvido com o sistema solvente álcool etílico absoluto: água purificada (60:40, v/v).
RESULTADOS E DISCUSSÃO
Desenvolvimento do método analítico
Conforme avaliação da solubilidade do EFZ frente a diferentes solventes, observou-se que este se apresenta como praticamente insolúvel ou insolúvel em água e em ácido clorídrico 0,1 M, conforme classificação da Farmacopeia Brasileira.22 Para o solvente acetonitrila, o fármaco apresentou-se facilmente solúvel, enquanto que para os solventes metanol e álcool etílico absoluto o fármaco se mostrou pouco solúvel.
Os resultados obtidos fornecem informações importantes sobre o comportamento do fármaco, possibilitando a escolha inicial dos solventes mais adequados para a completa solubilização do EFZ e que permitam a máxima estabilidade da solução, proporcionando a quantificação adequada do fármaco.
Para o método em desenvolvimento, utilizaram-se inicialmente metanol, álcool etílico e acetonitrila, como solventes iniciais de solubilização. Após a escolha destes solventes, foram realizadas varreduras utilizando várias combinações de solventes para a diluição das soluções, incluindo a possibilidade da adição de uma proporção de água. A proporção deste no sistema foi determinada de acordo com a estabilidade do EFZ em permanecer solúvel por 48 h em repouso, após completa solubilização. As combinações dos sistemas testados para a preparação das amostras estão descritas na Tabela 1.
As varreduras espectrofotométricas, realizadas para verificação dos sistemas que apresentavam as melhores absorções na faixa de comprimentos de onda entre 200 e 700 nm, demonstraram que todos os sistemas testados apresentaram absorções semelhantes no comprimento de 247 nm; com exceção do sistema acetonitrila/água, que apresentou uma absorção duas vezes maior aos outros sistemas na concentração de 10 μg mL-1. Portanto, devido à alta solubilização deste fármaco neste solvente e, consequentemente, da absorção elevada na faixa de concentração testada, esse sistema mostrou-se inadequado para o método.
Somado a este fato, para a escolha final do sistema a ser utilizado, levou-se em consideração a toxicidade e o custo do solvente, além da possível adição de uma proporção de água ao sistema, o que as facilita as leituras provenientes de estudos de preparação de complexos com ciclodextrinas, dispersões sólidas e ensaios de dissolução. Levando em consideração esses critérios, optou-se pelos solventes álcool etílico/sistema de diluição (álcool etílico:água, 60:40, v/v) para a primeira e segunda diluições, respectivamente (Figura 1).
Nas análises realizadas para a determinação do tipo e tempo de agitação na preparação das amostras, foi possível definir a ultrassonicação por 10 min como o método a ser padronizado, uma vez que este se apresentou como o mais eficiente para o método proposto (Tabela 2).
Validação do método analítico
Robustez
O método desenvolvido demonstrou ser robusto quanto à variação dos fabricantes de álcool etílico absoluto. De acordo com os resultados obtidos, o F calculado para esse parâmetro foi de 0,0218, inferior ao F tabelado (7,7086), sendo o CV de 0,28 e 0,14 para os fabricantes Dinâmica® e Vetec®, respectivamente. Com relação à avaliação da estabilidade das soluções no intervalo de 4 h a partir da preparação das amostras e da influência da luminosidade, obteve-se um F calculado de 2,1185 (F tabelado = 5,1922) e 4,1784 (F tabelado = 5,3176), respectivamente, demonstrando que não houve diferenças estatisticamente significativas entre os mesmos (Tabela 3).
Linearidade
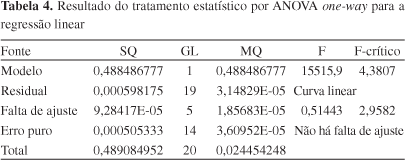
A análise de regressão linear dos mínimos quadrados apresentou um coeficiente de correlação de 0,99978, indicando linearidade dentro dos limites das concentrações estudadas, obtendo-se a equação da reta y = 0,0520x - 0,0099. Através da análise de variância (Tabela 4) pode-se testar a validação do método e a significância estatística da curva ajustada. De acordo os resultados, observou-se que o método é linear e não houve falta de ajustes para as médias estudadas, apresentando um valor de F de 2,9582, abaixo do valor crítico tabelado (4,3807).
Ainda como resultado da avaliação da linearidade, calcularam-se os LD e LQ. Os resultados encontrados foram de 0,21 para LD e 0,69 μg mL-1 para LQ.
Precisão
O método desenvolvido demonstrou ser preciso nos três níveis avaliados: repetitividade, precisão intermediária e reprodutibilidade. Para o parâmetro repetitividade os resultados obtidos apresentaram uma média de 100,32 e um CV de 1,35%, abaixo do valor máximo especificado.20
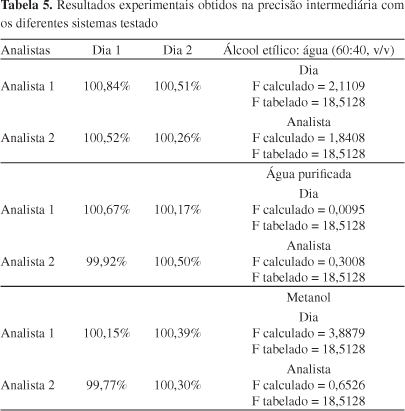
Na precisão intermediária foi demonstrado que o método é preciso para análises realizadas por analistas diferentes em um mesmo dia e em dias diferentes, estando a variação encontrada dentro dos limites especificados, pois através do tratamento estatístico foi demonstrado que os F calculados para as análises foram menores do que o F crítico (Tabela 5).
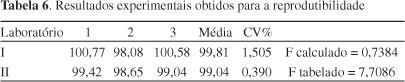
O método também se apresentou reprodutivo quando avaliado em diferentes laboratórios, utilizando equipamentos diferentes. Os resultados avaliados podem ser visualizados na Tabela 6.
Exatidão
A exatidão do método foi averiguada através da análise de três concentrações distintas (80, 100 e 120%), encontrando-se os resultados dentro dos limites especificados. Para este parâmetro, foi aplicado o teste t Student, o qual demonstrou que o t calculado para cada concentração foi de 0,8308; 0,8543 e 0,3523, respectivamente, sendo estes menores que o t tabelado (4,3026), comprovando que não houve diferença significativa entre as médias das três concentrações analisadas.
Especificidade
Para o método proposto, a solução amostra de EFZ foi contaminada com uma impureza, a zidovudina. Embora este fármaco possua absorção máxima no comprimento de onde de 270 nm,23 muito próximo ao comprimento em estudo (247 nm), não houve interferência na absorção do EFZ neste comprimento de onda e na concentração de 0,2 μg mL-1 de zidovudina. Esse valor encontra-se dentro do intervalo que compreende os LD e LQ da zidovudina, que são de 0,0394 e 0,0597 μg mL-1,23 respectivamente (Figura 2).
Absortividade molar (ε)
A absorbância da amostra foi de 0,555 para o solvente metanol e 0,318 para a água purificada, por meio da aplicação da equação M = m/mol x V; onde M é a concentração molar, m é a massa e V o volume. Conhecida a massa molar do EFZ (315,68 g mol-1), tem-se que a concentração molar do mesmo a 10 μg mL-1 é igual a 3,1677 x 10-5 mol. Aplicando-se a equação ε = Abs.M-1, calculou-se a absortividade molar do EFZ em água purificada (9028,63 M"1 cm"1) e metanol (17520,59 M"1 cm"1). Uma vez realizado o calculado da absortividade molar do fármaco no solvente de interesse, pode-se descobrir a concentração molar de qualquer amostra desconhecida solubilizada no solvente em questão.
Verificação da aplicabilidade da absortividade molar
Precisão
O método desenvolvido demonstrou ser preciso por meio da repetitividade e precisão intermediária para os dois solventes analisados. Na repetitividade, os resultados obtidos apresentaram um CV de 0,92% para a água purificada e 1,13% para o metanol. Na precisão intermediária foi demonstrado que o método é preciso para análises realizadas por analistas diferentes em um mesmo dia e em dias diferentes, sendo a variação enquadrada dentro dos limites especificados para ambos solventes (Tabela 5).
Especificidade
Para o método proposto, a solução amostra de EFZ nos dois solventes analisados foi contaminada com uma impureza, zidovudina, não havendo interferência na absorção do EFZ neste comprimento de onda.
Linearidade
Os resultados do estudo de linearidade estão agrupados na Tabela 7, bem como a média dos pontos estudados. A análise de regressão linear dos mínimos quadrados demonstrou um coeficiente de correlação de 0,9994 e 0,9939 para metanol e água, respectivamente, indicando a linearidade dentro dos limites das concentrações estudadas, obtendo-se a equação da reta y = 1,1107x + 0,0011 para o metanol e y = 0,9308x + 0,0081 para água. De acordo com os resultados, verificou-se que o método é linear e não houve falta de ajustes para as médias estudadas, apresentando um valor de F de 2,9582, abaixo do valor crítico tabelado de 4,3807.
Aplicando-se o método de comparação entre duas grandezas, no qual o módulo do valor teórico subtraído do valor prático, sendo o resultado dividido pelo valor teórico e multiplicando por cem, obtém-se em percentual o quão distante o resultado experimental está do modelo teórico. Para os valores avaliados na linearidade, para ambos solventes menores, a variação encontrada foi menor do que 5%, sendo, portanto, adequados analiticamente (Tabela 7).20
CONCLUSÃO
O método apresentado foi desenvolvido e validado segundo o ICH (ICH Q2A, 2005; ICH Q2B, 2005) e a ANVISA (RE n° 899, 2003), apresentou a confiabilidade requerida para um método analítico. Além de se mostrar como uma alternativa rápida, segura e de baixo custo quando comparado ao método por CLAE.
Paralelamente, a absortividade molar pode ser utilizada no setor de Desenvolvimento Analítico para facilitar o desenvolvimento de novos métodos, a investigação de desvios de qualidade e/ou adaptação dos mesmos. Por meio dos parâmetros utilizados, demonstraram-se as vantagens do uso dentro do limite de confiabilidade analítica.
Recebido em 29/11/09; aceito em 13/4/10; publicado na web em 20/7/10
- 1. Csajka, C.; Marzolini, C.; Fattinger, K.; Decosterd, L. A.; Fellay, J.; Telenti, A.; Biollaz, J.; Buclin, T.; Clin. Pharmacol. Ther. 2003, 20, 73.
-
2Brasil, Ministério da Saúde, Secretaria de Vigilância em Saúde, Programa Nacional de DST e Aids; Recomendações para terapia anti-retroviral em adultos infectados pelo HIV: manual de bolso, Ministério da Saúde: Brasília, 2008.
- 3. Montgomery, E. R.; Edmanson, A. L.; Cook, S. C.; Hovsepian, P. K.; J. Pharm. Biomed. Anal. 2001, 25, 267.
- 4. Mogatle, S.; Kanfer, I.; J. Pharm. Biomed. Anal. 2009, 49, 1308.
- 5. Martin, J.; Deslandes, G.; Dailly, E.; Renaud, C.; Reliquet, V.; Raffi, F.; Jolliet, P.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2009, 877, 3072.
- 6. Ramachandran, G.; Hemanth, A. K.; Swaminathan, S.; Venkatesan, P.; Kumaraswami, V.; Greenblatt, D. J.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2006, 835, 131.
- 7. Dailly, E.; Raffi, F.; Jolliet, P.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2004, 813, 353.
- 8. Matthews, C. Z.; Woolf, E. J.; Mazenko, R. S.; Haddix-Wiener, H.; Chavez-Eng, C. M.; Constanzer, M. L.; Doss, G. A.; Matuszewski, B. K.; J. Pharm. Biomed. Anal 2002, 28, 925.
- 9. Kappelhoff, B. S.; Alwin, H. R.; Huitema, D. R.; Beijnen, J. H.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2003, 792, 353.
- 10. Rezk, N. L.; Tidwell, R. R.; Kashuba, A. D. M.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2002, 774, 79.
- 11. Langmann, P.; Schirmer, D.; Väth, T.; Zilly, M.; Klinker, M. J.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2001, 755, 151.
- 12. Sarasa-Nacenta, M.; López-Púa, Y.; López-Cortés, L. F.; Mallolas, J.; Gatell, J. M.; Xavier, C.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2001, 763, 53.
- 13. Marzolini, C.; Telenti, A.; Buclin, T.; Biollaz, J.; Decosterd, L. A.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2000, 740, 43.
- 14. Veldkamp, A. I.; Heeswijk, R. P. G.; Meenhorst, P. L.; Mulder, J. W.; Lange, J. M. A.; Beijnen, J. H.; Hoetelmans, R. M. W.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 1999, 734, 55.
- 15. Soares, M. F. L. R.; Soares-Sobrinho, J. L.; Grangeiro-Júnior, S.; Silva, K. E. R.; Rolim-Neto, P. J.; Lat. Am. J. Pharm. 2008, 27, 688.
- 16. Galo, A. L.; Colombo, M. F.; Quim. Nova 2009, 32, 488.
- 17. Ribani, M.; Bottoli, C. B. G.; Collins, C. H.; Jardim, I. C. S. F.; Melo, L. F. C.; Quim. Nova 2004, 27, 771.
-
18International Conference on Harmonisation (ICH); Validation of Analytical Procedures: Definitions and Terminology, Q2A (CPMP/ICH/381/95), 1995.
-
19International Conference on Harmonisation (ICH); Validation of Analytical Procedures: Methodology, Q2B (CPMP/ICH/281/95), 1995.
-
20Brasil; Resolução RE nº899 de 29/5/2003; Diário Oficial da União, Brasília, DF, 02/06/2003, seção 1 - Agência Nacional de Vigilância Sanitária (ANVISA) aprova o Guia para validação de métodos analíticos e bioanalíticos.
- 21. Rocha, F. R. P.; Teixeira, L. S. G.; Quim. Nova 2004, 27, 807.
-
22Farmacopéia Brasileira, IV ed., Atheneu: São Paulo, 2003.
- 23. Randau, K. P.; Meira, J. L.; Braga, J. M. F.; Monteiro, D. B.; Rolim-Neto, P. J.; Acta Farm. Bonaerense 2005, 24, 104.
Datas de Publicação
-
Publicação nesta coleção
13 Jan 2011 -
Data do Fascículo
2010
Histórico
-
Recebido
29 Nov 2009 -
Aceito
13 Abr 2010