Resumo
Microwave synthesis is considered a breakthrough in the field of chemistry due to its benefits over conventional heating methods. We describe, for the first time, the total synthesis of phenytoin adapted for a microwave reactor, proposed as an experiment for undergraduate courses. When carried out with microwaves, the synthetic route provided phenytoin in 6 min and 30 s, while for conventional heating the total time was 170 min. The experiment also involves rapid identification of compounds by TLC, ¹H NMR, FT-IR and melting points determination.
drug synthesis; phenytoin; microwave-assisted organic synthesis
drug synthesis; phenytoin; microwave-assisted organic synthesis
EDUCAÇÃO
Síntese da fenitoína assistida por micro-ondas: proposta de aula prática para alunos de graduação
Microwave-assisted phenytoin synthesis: proposed practical class for undergraduate students
Eduardo L. Konrath* * e-mail: edukonrath@yahoo.com.br ; Maristela Piedade; Vera Lucia Eifler-Lima
Departamento de Produção de Matéria-Prima, Faculdade de Farmácia, Universidade Federal do Rio Grande do Sul, Av. Ipiranga, 2752, 90610-000 Porto Alegre - RS, Brasil
ABSTRACT
Microwave synthesis is considered a breakthrough in the field of chemistry due to its benefits over conventional heating methods. We describe, for the first time, the total synthesis of phenytoin adapted for a microwave reactor, proposed as an experiment for undergraduate courses. When carried out with microwaves, the synthetic route provided phenytoin in 6 min and 30 s, while for conventional heating the total time was 170 min. The experiment also involves rapid identification of compounds by TLC, 1H NMR, FT-IR and melting points determination.
Keywords: drug synthesis; phenytoin; microwave-assisted organic synthesis.
INTRODUÇÃO
Um dos primeiros fármacos com atividade antiepilética introduzidos na terapêutica, ainda no início do século XX foi o fenobarbital (1), produzido pela Bayer e que demonstrou eficácia como anticonvulsivante.1 Alguns anos mais tarde, Merrit e Putnam2 testaram alguns derivados de hidantoína em um modelo de epilepsia induzida por eletrochoques em gatos e verificaram que a fenitoína (5,5-difenil-2,4-imidazolidinediona) (2) foi a substância mais potente dentre as ensaiadas, sendo dotada de efeito anticonvulsivante, mas sem o efeito sedativo apresentado pelos barbitúricos. O mecanismo de ação da fenitoína se baseia na inibição dos canais de sódio sensíveis à voltagem, com a consequente estabilização das membranas neuronais,3 de forma que ainda hoje ela é considerada como o fármaco de escolha no tratamento da epilepsia, especificamente de convulsões parciais e tônico-clônicas, pela sua boa eficácia apresentada.4
A fenitoína foi primeiramente sintetizada por Michael,5 mas sua estrutura foi corretamente elucidada somente alguns anos depois por Biltz,6 que descreveu pela primeira vez sua obtenção a partir da condensação da benzila através de uma catálise básica com ureia, motivo pelo qual essa reação é conhecida como síntese de Biltz.7 Algumas décadas mais tarde, Dumavant e colaboradores8 reinvestigaram esta rota sintética e verificaram que a síntese de Biltz ocorre via um rearranjo benzílico e que, além disso, esta reação promovia uma obtenção concomitante e em proporções iguais tanto de fenitoína quanto do aduto 3a,7a-difenilglicorudeída (3). Os mesmos autores também verificaram que a proporção 1:2 dos reagentes benzila e ureia a ser empregada nesta reação seria a ideal, a fim de se reduzir a formação deste produto secundário.
Nesse caso, relatos na literatura indicam que o uso de um sistema bifásico, como uma solução aquosa de KOH/n-BuOH e PEG600 como fase catalítica de transferência, é capaz de reduzir significativamente a formação da glicorudeída 3, além de promover um aumento no rendimento da fenitoína.9 Mais recentemente, Muccioli e colaboradores10 reportaram uma adaptação da síntese de Biltz com o uso de micro-ondas, permitindo a formação tanto de hidantoínas quanto de tio-hidantoínas de maneira mais rápida e limpa, características estas comuns de reações por micro-ondas. Neste estudo, o uso de diferentes solventes foi primeiramente testado, de maneira que os sistemas DMSO e dioxano/H2O (2:5 v:v) promoveram um maior rendimento e seletividade para a reação.
Um dos principais objetivos das aulas experimentais da disciplina de Síntese Orgânica de Fármacos11 é a obtenção de fármacos em poucas etapas e sem acarretar grandes perdas em termos de rendimento quanto ao produto final devido a interrupções temporárias.12 Nesse sentido, propomos a obtenção da fenitoína através da síntese de Biltz para aula prática, usando como fonte de aquecimento um reator de micro-ondas em todas as etapas da rota. Para comparação serão utilizados os rendimentos obtidos através do método clássico, com aquecimento convencional. Esta prática nos permite familiarizar os estudantes em técnicas sintéticas e também introduzir os conceitos de síntese orgânica assistida por micro-ondas (microwave assisted organic synthesis - MAOS).
RESULTADOS E DISCUSSÃO
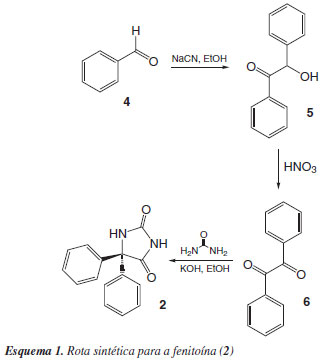
A síntese de Biltz, que leva à obtenção da fenitoína, é uma das mais comuns realizadas nas aulas experimentais de Síntese Orgânica de Fármacos, em cursos de Graduação de Farmácia.13 As aulas práticas foram planejadas de forma que os alunos possam rever os conceitos básicos introduzidos nas disciplinas pré-requisitos (Química Orgânica Experimental e Química Farmacêutica), aprender o escopo da rota sintética planejada, juntamente com os mecanismos e tipos de reações que ocorrem nas etapas da mesma, bem como as limitações das reações, caracterização dos intermediários-chave e do produto final, e a análise farmacêutica do fármaco obtido. Adicionalmente, os estudantes aplicam os conhecimentos prévios de infravermelho (IV) e ressonância magnética nuclear de hidrogênio (RMN-1H) para acompanhar a evolução da síntese através da interpretação dos espectros do material de partida, benzaldeído (4), dos intermediários benzoína (5) e benzila (6) e do produto final (2). Dentre os métodos adicionais para a análise dos produtos obtidos, estão a realização de análises cromatográficas (CCD) com o uso de amostras autênticas obtidas e ponto de fusão (Pf), por comparação com dados previamente descritos na literatura.
Os alunos da disciplina de Síntese Orgânica de Fármacos realizam a síntese da fenitoína em três etapas empregando aquecimento convencional (aquecimento por convecção) e, para tanto, necessitam de 3 aulas práticas, sendo cada etapa elaborada em uma aula, conforme esquema demonstrado na Figura 1. O objetivo deste trabalho é de introduzir os alunos à prática da síntese em micro-ondas, de forma que a rota proposta leve os alunos a realizarem todas as etapas em uma aula apenas (ca de 10 min). As reações realizadas sob irradiação das micro-ondas são mais rápidas, conferem seletividade e produtos com maior grau de pureza, uma vez que o aquecimento proporcionado é rápido e uniforme, e vem ganhando cada vez mais espaço no universo da Química.14 O emprego de micro-ondas para reações em aulas práticas para alunos de Graduação também vem sendo bastante descrito devido a sua fácil implementação, tornando as aulas mais simples e rápidas.15 Neste trabalho, propomos o uso de um reator de micro-ondas, feito especificamente para reações orgânicas, para a obtenção da fenitoína.
A primeira etapa da rota sintética envolve a formação de benzoína, uma alfa-hidroxicetona, através de uma condensação a partir do benzaldeído, com o uso de um catalisador adequado. O procedimento clássico desta etapa envolve uma reação promovida em meio alcalino com a presença de íons cianeto, formando um carbânion estável com um grupamento nitrila.16 Assim, devido à toxicidade da reação e de seus reagentes, essa reação é sempre realizada sob capela de exaustão, e o resíduo gerado é tratado com sulfato de ferro (II), com o intuito de gerar um complexo menos tóxico e passível de tratamento posterior, o hexacianoferrato (II) de ferro (III).17 Na busca de catalisadores menos tóxicos, como alternativas já descritas na literatura para os íons cianeto, estão a tiamina,18 bem como sais de tiazol em meio alcalino,19 carbenos heteroazólios 20 e metalofosfitos. 21 Também se encontram descritas reações de condensação para a formação de benzoína sem o uso de solventes e com o uso de micro-ondas, empregando-se líquidos iônicos de sais de imidazol.22
Com base nesses dados, foram desenvolvidos dois métodos com o uso de micro-ondas para a primeira etapa da reação, sendo um a adaptação do protocolo já estabelecido do método clássico com os mesmos reagentes, e o segundo empregando-se uma base forte como agente promotor da reação;23 no nosso caso, utilizamos o hidróxido de potássio. O maior rendimento obtido com o uso de MAOS foi para o primeiro método utilizado (86%), enquanto que a segunda reação proporcionou um rendimento de 77%; cabe salientar que ambos os rendimentos foram superiores àquele obtido pela reação com o aquecimento clássico (≈ 69%).
A oxidação da benzoína à sua respectiva alfa-dicetona, a benzila, é um método relativamente simples e amplamente descrito na literatura com o uso de diversos agentes oxidantes, como ácido nítrico,24 solução de Fehling,25 nitrato de tálio (III),26 acetato de nitrato-cobre (II) e bismuto (III),27 sais de cromo com alumina ácida,28 dentre outros. Mais recentemente, foi relatada uma reação de oxidação para a benzoína empregando-se hidreto de sódio,29 resultando em um rendimento elevado e sem o emprego de reagentes tóxicos, numa busca por um processo sintético mais limpo e seguro do ponto de vista ambiental. A reação correspondente à etapa de oxidação da benzoína realizada em aula prática pelo método convencional envolve o uso de ácido nítrico concentrado, devendo ser realizado em capela de exaustão devido à formação de vapores nitrosos tóxicos. Mesmo tratando-se de uma etapa com riscos em potencial, devido às características corrosivas e tóxicas do ácido nítrico, o resíduo gerado nesta reação pode ser facilmente descartado através de alcalinização posterior do filtrado ácido obtido após a separação da benzila.
O procedimento para as reações em micro-ondas nesta etapa foi realizado visando um método alternativo para o uso de um ácido fumegante concentrado. Nesse caso, nossa primeira tentativa envolveu a utilização do reagente oxidante dióxido de manganês com suporte em sílica (MnO2/SiO2), conforme experimentos previamente descritos usando micro-ondas e sem solventes.30 O rendimento obtido para esta reação foi baixo (≈ 36%), de maneira que a segunda tentativa realizada foi com o CuSO4/Al2O3,31 proporcionando um rendimento de 64%, superior à reação anterior mas ainda menor quando comparado àquele obtido pelo método convencional (88,5%).
A benzila assim obtida é empregada como ponto de partida para a última etapa do processo de síntese, com a consequente obtenção da fenitoína. Este produto final é gerado através de uma condensação com a ureia em meio alcoólico alcalino, conforme o procedimento clássico da síntese de Biltz. O método empregado em micro-ondas para esta etapa foi, do mesmo modo, uma adaptação do método convencional, proporcionando um rendimento superior (89%) quando comparado àquele obtido por aquecimento por convecção (74,5%).
A formação dos produtos e a evolução das reações puderam ser acompanhadas por CCD e posterior visualização em câmara de UV ou revelação com iodo. As análises dos espectros de IV e RMN-1H permitiram a identificação inequívoca dos compostos que se apresentavam puros.
PARTE EXPERIMENTAL
Benzaldeído (Sigma-Aldrich), cianeto de sódio, sulfato cuproso, alumina neutra, sílica, dióxido de manganês (Merck), ácido nítrico, hidróxido de potássio e ureia (Vetec) foram adquiridos comercialmente. O etanol e o benzaldeído foram previamente destilados sob pressão atmosférica antes de serem empregados.
Os espectros vibracionais na região do infravermelho foram registrados em espectrofotômetro Perkin-Elmer Spectrum BXII em pastilhas de KBr. Os espectros de RMN de hidrogênio foram feitos em espectrofotômetro Brucker INOVA-300 MHz, usando tetrametilsilano (TMS) como padrão interno. O reator de micro-ondas foi da marca CEM Discover, modelo 908010, utilizado dentro de uma capela e operado na potência de 100 W em todas as reações.
Os pontos de fusão não corrigidos foram medidos em aparelho tipo Kofler WME, termômetro -10 a 300 ºC, aferido com ácido benzoico, Merck (Pf: 122,6 a 123,1 ºC).
Procedimento
A síntese da fenitoína foi realizada em três etapas. Os produtos obtidos em cada uma das etapas intermediárias foram secos em dessecador por uma semana, durante o período de intervalo entre cada aula prática. Para a obtenção dos produtos pelo método clássico, as reações foram realizadas em balões de fundo redondo com duas bocas, sendo o agitador magnético introduzido após a adição dos reagentes. Nas reações com o uso do reator de micro-ondas, foi empregado o modo de agitação média. Os demais parâmetros adotados nas reações com o uso do reator de micro-ondas, bem como os rendimentos obtidos para ambos os métodos, estão descritos na Tabela 1.
Etapa 1: Preparação da benzoína (5), reação de condensação
Método clássico: em balão de fundo redondo de 100 mL, foram adicionados benzaldeído puro (4) (4 g, 37 mmol), solução de cianeto de sódio 10% m/v (5 mL) e etanol (7 mL). A mistura reacional foi acoplada a um condensador de refluxo e aquecida com agitação magnética durante 30 min. Após esse período, o balão foi resfriado em banho de gelo, ocorrendo a formação do precipitado, que foi filtrado sob vácuo. O produto foi, em seguida, seco em dessecador e obtido na forma de cristais amarelo-limão (2,75 g). Pf.: 134-136 ºC (lit.: 137 ºC)32; CCD: Rf.: 0,65 com eluente tolueno/acetato de etila/etanol (9:0,5:0,5), seguida de visualização em câmara de ultravioleta; IV (KBr, cm-1): vmáx: 3400 (OH), 1677 (C=O), 753 (C-H, anel aromático monossubtituído); RMN (300 MHz, CDCl3): H 4,5 (s, 1H, OH); 5,9 (s, 1H, C-H), 7-8 (m, 10H, Ar-H). Os espectros obtidos correspondem aos do catálogo de padrões internacionais (medidos com amostras autênticas), SDBS nº 1700.33
Método assistido por micro-ondas: para a primeira reação, foram adicionados em um tubo de reação benzaldeído puro (0,1 mL, 1 mmol), solução de cianeto de sódio 10% m/v (0,125 mL) e etanol (0,17 mL). Na segunda reação, foram adicionados benzaldeído puro (0,1 mL, 1 mmol), KOH (s) pulverizado (0,168 g, 3 mmol) e metanol (0,13 mL). Depois de completada a reação, monitorada por CCD (mesmas condições citadas anteriormente), o produto foi filtrado sob vácuo e seco em dessecador, fornecendo 0,09 e 0,08 g de benzoína, respectivamente.
Etapa 2: Preparação da benzila (6), reação de oxidação
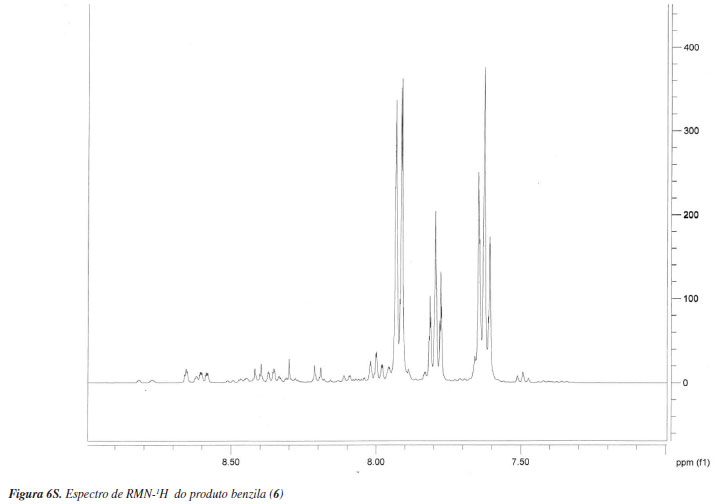
Método clássico: em um balão de fundo redondo de 100 mL, foram aquecidos benzoína (5) (7 g, 33 mmol) com HNO3 concentrado (70 mL) adicionado em uma única porção, sob agitação constante, sendo o sistema acoplado a um condensador de refluxo durante 60 min com agitação magnética, até cessar o desprendimento de vapores nitrosos, sob capela de exaustão. Após esse período, a mistura reacional foi vertida sobre uma mistura de 100 mL de água destilada com gelo picado. O produto foi, em seguida, filtrado sob vácuo e seco em dessecador, sendo obtido na forma de cristais amarelo pálidos (6,19 g). Pf.: 94-96 ºC (lit.: 95 ºC)32; CCD: Rf.: 0,57 com eluente tolueno/acetato de etila/etanol (9:0,5:0,5), seguida de visualização em câmara de ultravioleta; IV (KBr, cm-1): vmáx: 1660 (C=O), 1592, 1580, 1450 (C=C, anel aromático); RMN (300 MHz, CDCl3): H 7,6 (t, 4H, Ar-H); 7,8 (t, 2H, Ar-H); 7,9 (t, 4H, Ar-H). Os espectros obtidos correspondem aos do catálogo de padrões internacionais (medidos com amostras autênticas), SDBS nº 1700.33
Método assistido por micro-ondas: para a primeira reação, foram utilizados benzoína (0,212 g, 1 mmol) e, como agente oxidante, a mistura MnO2/SiO2 (1,5 g), preparada através da pulverização de ambos os reagentes em um gral, na proporção 1:2. Para a segunda reação, foram adicionados em um tubo de reação benzoína (0,212 g, 1 mmol) e o agente oxidante CuSO4/Al2O3 (1,5 g, 0.85 mmol de CuSO4.5H2O), preparado impregnando-se alumina neutra (Al2O3) com solução aquosa de CuSO4 8% m/v, ambos na proporção 1:2 (25 g de Al2O3 para 50 mL de solução de CuSO4.5H2O a 8%), durante 12 h com agitador magnético. Em seguida, a alumina foi filtrada e seca em estufa a 45 ºC durante 12 h antes de ser empregada. Depois de completada a reação de oxidação, com monitoramento por CCD (mesmas condições citadas anteriormente), o produto foi extraído com diclorometano (3 x 1 mL) e o solvente evaporado. Após secagem em dessecador, o produto foi então purificado através de CCD preparativa, com a fase móvel tolueno/acetato de etila/etanol (9:0,5:0,5), sendo então novamente seco em dessecador. As quantidades de benzila obtidas foram de 0,07 e 0,135 g para a primeira e segunda reações, respectivamente.
Etapa 3: Preparação da fenitoína ( 2 )
Método clássico: em um balão de fundo redondo de 100 mL, foram adicionados a benzila (6) (5,25 g, 25 mmol), ureia (2,25 g, 37 mmol), solução de KOH 66% m/v (12,5 mL) e etanol (75 mL). O sistema foi acoplado a um condensador de refluxo e aquecido durante 60 min com agitação magnética. Após esse período, a mistura reacional foi transferida para um béquer e foi adicionado igual volume de água destilada gelada. A mistura foi então filtrada sob vácuo e (2), que estava na forma de sal potássico solúvel, sofreu precipitação completa durante a adição de uma solução de HCl 20% v/v. A fenitoína assim gerada foi filtrada e seca em dessecador, sendo obtida na forma de cristais de cor esbranquiçada (3,91 g). Pf.: 294-296 ºC (lit.: 295-298 ºC);32 CCD: Rf.: 0,34 com eluente tolueno/acetato de etila/etanol (9:0,5:0,5), seguida de visualização em câmara de ultravioleta; IV (KBr, cm-1): vmáx: 3450 e 3420 (N-H); 1720 e 1769 (C=O); RMN (300 MHz, CDCl3): δH 7,4 (m, 10H, Ar-H); 9,3 (s, 1H, N-H); 11,2 (s, 1H, N-H). Os espectros obtidos correspondem aos do catálogo de padrões internacionais (medidos com amostras autênticas), SDBS nº 7403.33
Método assistido por micro-ondas: para a reação, foram adicionados em um tubo de reação a benzila (0,262 g, 1,2 mmol), ureia (0,1 g, 1,5 mmol), solução de KOH 66% m/v (0,5 mL) e etanol (3,5 mL). Depois de completada a reação, monitorada por CCD (mesmas condições citadas anteriormente), o meio reacional foi também acidificado com solução de HCl 20% v/v até precipitação completa do produto. A fenitoína foi então filtrada sob vácuo e os cristais obtidos secos em dessecador (0,23 g) (Esquema 1).
CONCLUSÕES
Os experimentos sequenciais realizados pelos alunos forneceram o fármaco fenitoína com grau de pureza satisfatório, tanto pelo método clássico com o uso de aquecimento convencional, quanto com o uso de reator de micro-ondas. As reações realizadas com micro-ondas forneceram a fenitoína em 6,5 min, enquanto que em aquecimento convencional foram necessários 170 min. As atividades interdisciplinares envolvendo síntese orgânica, análise espectroscópica, cromatografia e controle de qualidade contribuíram no processo ensino-aprendizagem na disciplina de Síntese Orgânica de Fármacos. Cabe salientar que esta é a primeira descrição da síntese total do fármaco fenitoína empregando-se MAOS, uma vez que os dados existentes na literatura conduzem apenas a algumas das etapas isoladamente com o uso de irradiação por micro-ondas. No final das aulas realizadas, os alunos foram capazes de relacionar adequadamente os conceitos teóricos e práticos durante a apresentação dos seminários, bem como na realização das provas teórico-práticas, comparando criticamente seus resultados com os demais grupos.
MATERIAL SUPLEMENTAR
Está disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS
À técnica J. Espíndola, do Instituto de Química - UFRGS, pela gentil realização dos espectros de RMN-1H, bem como à CAPES e ao CNPq pelo apoio financeiro concedido.
REFERÊNCIAS E NOTAS
1. Schmidt, D.; Drug Trial in Epilepsy,a Physician's Guide, Martin Dunitz: London, 1998; Guerreiro, C. A. M.; J. Epilepsy Clin. Neurophysiol. 2006,12, suppl. 1,18.
2. Putnam, T. J.; Merrit, H. H.; Science 1937,85,525.
3. Bruni, J. Em Antiepileptic drugs; Levy, R. H.; Meldrum, B. S., eds.; 4th ed., Raven Press: New York, 1995.
4. Del Negro, A.; Dantas, C. R.; Zanardi, V.; Montenegro, M. A.; Cendes, F.; Arq. Neuro-Psiquiatr. 2000,58,276.
5. Michael, A.; Am. Chem. J. 1887,9,219.
6. Biltz, H.; Chem. Ber. 1908,41,1379.
7. Hayward, R. C. J.; J. Chem. Educ. 1983,60,512.
8. Dunnavant, W. R.; James, F. L.; J. Am. Chem. Soc. 1956,78,2740.
9. Poupaert, J. H.; De Keyser, J. L.; Vandervorst, D.; Dumond, P.; Bull. Soc. Chim. Belg. 1984,93,493.
10. Muccioli, G. G.; Poupaert, J. H.; Wouters, J.; Norberg, B.; Poppitz, W.; Scriba, G. K. E.; Lambert, D. M.; Tetrahedron 2003,59,1301.
11 A disciplina de Síntese Orgânica de Fármacos (FAR01109) é ministrada para o curso de Farmácia da Universidade Federal do Rio Grande do Sul, ênfase em Indústria de Medicamentos, compreendendo tanto aulas teóricas como práticas. Esta disciplina aborda a síntese de fármacos, como a fenitoína aqui apresentada. Entretanto, esta aula também pode ser perfeitamente aplicada em outras disciplinas tanto dos cursos de Química como de Farmácia.
12. Borges, A. D. L.; Del Ponte, G.; Federman Neto A.; Carvalho, I.; Quim. Nova 2005,28,727.
13. Eicher, T.; Roth, H. J.; Synthese, Gewinnung und Charakterisierung von Arzneistoffen: Ein Pratikumsbuch, Georg Thieme Verlag Stuttgard: New York, 1986; Soares, B. G.; Souza, N. A.; Pires, D. X.; Química Orgânica: Teoria e Técnicas de Preparação, Purificação e Identificação de Compostos Orgânicos, Ed. Guanabara S.A.: Rio de Janeiro, 1988.
14. Raveglia, L. F.; Giardina, G. A. M.; Future Med. Chem. 2009,1,1019; Teixeira, E. F.; dos Santos, A. P. B.; Bastos, R. S.; Pinto, A.; Kümmerle, A. E.; Coelho, R. R.; Quim. Nova 2010,33,1603.
15. Rosini, F.; Nascentes, C. C.; Nóbrega, J. A.; Quim. Nova 2004,27,1012; Graebin, C. S.; Eifler-Lima, V. L.; Quim. Nova 2005,28,73; de Souza, R. O.; Miranda, R. S.; Quim. Nova 2011,34,497.
16. Bausch, C. C.; Johnson, J. S.; J. Org. Chem. 2004,69,4283; Bausch, C. C.; Johnson, J. S.; Adv. Synth. Catal. 2005,347,1207.
17. Picot, A.; Grenouillet, P.; La Securité en Laboratoire de Chimie et de Biochimie, Tec & Doc Lavoisier: Paris, 1992.
18. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Engel, R. G.; Química Orgânica Experimental: Técnicas de Pequena Escala, 2ª ed., Bookman: Porto Alegre, 2009.
19. Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowiski, E. J. J.; Reider, P. J.; J. Am. Chem. Soc. 2001,123,9696; Tachibana, P.; Kihara, N.; Takata, T.; J. Am. Chem. Soc. 2004,126,3438.
20. Chan, A.; Scheidt, K. A.; Org. Lett. 2005,7,905.
21. Linghu, X.; Potnick, J.; Johnson, J.; J. Am. Chem. Soc. 2004,126,3070.
22. Xu, L. W.; Gao, Y.; Yin, J. J.; Li, L.; Xia, C. G.; Tetrahedron Lett. 2005,46,5317; Estager, J.; Lévêque, J. M.; Turgis, R.; Draye, M.; J. Mol. Catal. A 2006,256,261; Estager, J.; Lévêque, J. M.; Turgis, R. ; Draye, M.; Tetrahedron Lett. 2007,48,755.
23. March, J.; Advanced Organic Chemistry, 4th ed., Willey Interscience: New York, 1992.
24. Zinin, N.; Ann. Chem. Pharm. 1940,34,186.
25. Buck, J. S.; Jenkins, S. S.; J. Am. Chem. Soc. 1929,51,2163.
26. McKillop, A.; Swann, B. P.; Ford, M. E.; Taylor, E. C.; J. Am. Chem. Soc. 1973,95,3641.
27. Tymonko, S. A.; Nattier, B. A.; Mohan, R. S.; Tetrahedron Lett. 1999,40,7657.
28. Sahu, A.; Rawal, M. K.; Sharma, V. K.; Parashar, B.; Int. J. ChemTech. Res. 2009,1,1087.
29. Joo, C.; Kang, S.; Kim, S. M.; Han, H.; Yang, J. W.; Tetrahedron Lett. 2010,51,6006.
30. Varma, R. S.; Saini, R. K.; Dahiya, R.; Tetrahedron Lett. 1997,45,7823.
31. Varma, R. S.; Kumar, D.; Dahiya, R.; J. Chem. Res. 1998,S,324.
32. The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals, 12th ed.; Rahway: New York, 1996.
33. Saito, T.; Hayamizu, K.; Yanagisawa. M.; Yamamoto, O.; Wasada, N.; Someno, K.; Kinugasa, S.; Tanabe, K.; Tamura, T.; Tanabe, K.; Hiraishi, J.; NIST - National Institute of Standards, AIST - Institute of Advanced Industrial Science and Technology, SDBS - Integrated Spectral Data Base System for Organic Compounds, disponível em http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/cre_index.cgi?lang=eng, acessada em Março 2012.
Recebido em 27/3/12; aceito em 27/4/12; publicado na web em 3/8/12
- 1. Schmidt, D.; Drug Trial in Epilepsy,a Physician's Guide, Martin Dunitz: London, 1998;
- Guerreiro, C. A. M.; J. Epilepsy Clin. Neurophysiol. 2006,12, suppl. 1,18.
- 2. Putnam, T. J.; Merrit, H. H.; Science 1937,85,525.
- 3. Bruni, J. Em Antiepileptic drugs; Levy, R. H.; Meldrum, B. S., eds.; 4th ed., Raven Press: New York, 1995.
- 4. Del Negro, A.; Dantas, C. R.; Zanardi, V.; Montenegro, M. A.; Cendes, F.; Arq. Neuro-Psiquiatr. 2000,58,276.
- 5. Michael, A.; Am. Chem. J. 1887,9,219.
- 6. Biltz, H.; Chem. Ber 1908,41,1379.
- 7. Hayward, R. C. J.; J. Chem. Educ 1983,60,512.
- 8. Dunnavant, W. R.; James, F. L.; J. Am. Chem. Soc. 1956,78,2740.
- 9. Poupaert, J. H.; De Keyser, J. L.; Vandervorst, D.; Dumond, P.; Bull. Soc. Chim. Belg. 1984,93,493.
- 10. Muccioli, G. G.; Poupaert, J. H.; Wouters, J.; Norberg, B.; Poppitz, W.; Scriba, G. K. E.; Lambert, D. M.; Tetrahedron 2003,59,1301.
- 12. Borges, A. D. L.; Del Ponte, G.; Federman Neto A.; Carvalho, I.; Quim. Nova 2005,28,727.
- 13. Eicher, T.; Roth, H. J.; Synthese, Gewinnung und Charakterisierung von Arzneistoffen: Ein Pratikumsbuch, Georg Thieme Verlag Stuttgard: New York, 1986;
- Soares, B. G.; Souza, N. A.; Pires, D. X.; Química Orgânica: Teoria e Técnicas de Preparação, Purificação e Identificação de Compostos Orgânicos, Ed. Guanabara S.A.: Rio de Janeiro, 1988.
- 14. Raveglia, L. F.; Giardina, G. A. M.; Future Med. Chem 2009,1,1019;
- Teixeira, E. F.; dos Santos, A. P. B.; Bastos, R. S.; Pinto, A.; Kümmerle, A. E.; Coelho, R. R.; Quim. Nova 2010,33,1603.
- 15. Rosini, F.; Nascentes, C. C.; Nóbrega, J. A.; Quim. Nova 2004,27,1012;
- Graebin, C. S.; Eifler-Lima, V. L.; Quim. Nova 2005,28,73;
- de Souza, R. O.; Miranda, R. S.; Quim. Nova 2011,34,497.
- 16. Bausch, C. C.; Johnson, J. S.; J. Org. Chem 2004,69,4283;
- Bausch, C. C.; Johnson, J. S.; Adv. Synth. Catal 2005,347,1207.
- 17. Picot, A.; Grenouillet, P.; La Securité en Laboratoire de Chimie et de Biochimie, Tec & Doc Lavoisier: Paris, 1992.
- 18. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Engel, R. G.; Química Orgânica Experimental: Técnicas de Pequena Escala, 2Ş ed., Bookman: Porto Alegre, 2009.
- 19. Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowiski, E. J. J.; Reider, P. J.; J. Am. Chem. Soc. 2001,123,9696;
- Tachibana, P.; Kihara, N.; Takata, T.; J. Am. Chem. Soc. 2004,126,3438.
- 20. Chan, A.; Scheidt, K. A.; Org. Lett 2005,7,905.
- 21. Linghu, X.; Potnick, J.; Johnson, J.; J. Am. Chem. Soc 2004,126,3070.
- 22. Xu, L. W.; Gao, Y.; Yin, J. J.; Li, L.; Xia, C. G.; Tetrahedron Lett. 2005,46,5317;
- Estager, J.; Lévêque, J. M.; Turgis, R.; Draye, M.; J. Mol. Catal. A 2006,256,261;
- Estager, J.; Lévêque, J. M.; Turgis, R. ; Draye, M.; Tetrahedron Lett 2007,48,755.
- 23. March, J.; Advanced Organic Chemistry, 4th ed., Willey Interscience: New York, 1992.
- 24. Zinin, N.; Ann. Chem. Pharm. 1940,34,186.
- 25. Buck, J. S.; Jenkins, S. S.; J. Am. Chem. Soc. 1929,51,2163.
- 26. McKillop, A.; Swann, B. P.; Ford, M. E.; Taylor, E. C.; J. Am. Chem. Soc 1973,95,3641.
- 27. Tymonko, S. A.; Nattier, B. A.; Mohan, R. S.; Tetrahedron Lett. 1999,40,7657.
- 28. Sahu, A.; Rawal, M. K.; Sharma, V. K.; Parashar, B.; Int. J. ChemTech. Res 2009,1,1087.
- 29. Joo, C.; Kang, S.; Kim, S. M.; Han, H.; Yang, J. W.; Tetrahedron Lett 2010,51,6006.
- 30. Varma, R. S.; Saini, R. K.; Dahiya, R.; Tetrahedron Lett 1997,45,7823.
- 31. Varma, R. S.; Kumar, D.; Dahiya, R.; J. Chem. Res. 1998,S,324.
- 32. The Merck Index: An Encyclopedia of Chemicals, Drugs and Biologicals, 12th ed.; Rahway: New York, 1996.
- 33. Saito, T.; Hayamizu, K.; Yanagisawa. M.; Yamamoto, O.; Wasada, N.; Someno, K.; Kinugasa, S.; Tanabe, K.; Tamura, T.; Tanabe, K.; Hiraishi, J.; NIST - National Institute of Standards, AIST - Institute of Advanced Industrial Science and Technology, SDBS - Integrated Spectral Data Base System for Organic Compounds, disponível em http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/cre_index.cgi?lang=eng, acessada em Março 2012.
Datas de Publicação
-
Publicação nesta coleção
05 Out 2012 -
Data do Fascículo
2012
Histórico
-
Recebido
27 Mar 2012 -
Aceito
27 Abr 2012