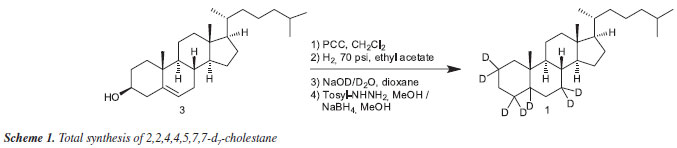
Abstract
A short and efficient synthesis of heptadeuterated 2,2,4,4,5,7,7-d7-cholestane (1) from cholesterol (3) is described. The deuterated material will be useful for the analysis of different sources of petroleum in analytical geochemistry laboratories as internal standard for quantification of steranes via gas chromatography-mass spectrometry (GC-MS).
total synthesis; steroids; deuterated standard
ARTIGO
Straightforward synthesis of 2,2,4,4,5,7,7-d7-cholestane: a new deuterated standard in petroleum analysis
Maicon Guerra de Miranda* * e-mail: maicon_iq@yahoo.com ; Andre Luis Mazzei Albert; Jari Nobrega Cardoso; Rosangela Sabbatini Capella Lopes; Claudio Cerqueira Lopes
Instituto de Química, Universidade Federal do Rio de Janeiro, Avenida Athos da Silveira Ramos, 149 CT, Bloco A, S.508, 21941-909 Cidade Universitária, Rio de Janeiro - RJ, Brasil
ABSTRACT
A short and efficient synthesis of heptadeuterated 2,2,4,4,5,7,7-d7-cholestane (1) from cholesterol (3) is described. The deuterated material will be useful for the analysis of different sources of petroleum in analytical geochemistry laboratories as internal standard for quantification of steranes via gas chromatography-mass spectrometry (GC-MS).
Keywords: total synthesis; steroids; deuterated standard.
INTRODUCTION
The identification of organic structures in the environment can be very complex1 with various processes frequently acting to modify structures and/or distributions of key compounds used as biomarkers. In some situations, examination of the relative distributions of biomarkers can be used as a method of fingerprinting and can be sufficient in providing the necessary answers. Unfortunately, this is frequently not the case, notably for the trace concentrations common in very mature petroleum samples.2 Other examples include the recognition of mixed oil-sources with very similar biomarker profiles, found both in exploration and oil-spill studies,3 and the correct interpretation of the changes in organic composition resulting from laboratory simulation experiments with geological materials by heating to understand petroleum generation, transformation or migration.4 In such cases, absolute quantification can be mandatory and availability of a standard of the compound of interest or a structurally related analog is essential. Unfortunately, the necessary standard is frequently not available, notably for complex structures. This problem is aggravated by the fact that, even when commercially available, such standards are extremely expensive for routine use, some easily costing over a thousand dollars for quantities in the milligram range.
The main objective of this work was to develop a simple synthetic scheme to prepare deuterated cholestanes (Scheme 1), frequently used as standards for quantification of steranes, biomarkers of great interest in petroleum and petroleum-related samples, in high yield and purity, using readily available reagents and starting materials. The synthesis should be simple enough to be performed by chemists with basic synthetic training in order to be useful, as an alternative, to the purchase of expensive commercial standards.
RESULTS AND DISCUSSION
Our synthetic route (Scheme 2) starts with the oxidation of cholesterol (3)thane suspension in the presence of cholesterol (3) to simultaneously promote two chemoselective, oxidation reactions, to furnish the product 4-cholesten-3,6-dienone (4) at a yield of 83%. Compound (4) was then submitted to catalytic hydrogenation conditions5 at 70 psi of H2 with 10% Pd-C and ethyl acetate as solvent for 40 hours in a Parr Apparatus, giving the corresponding 3,6-cholestandione (5) in quantitative yield.
Various procedures described in the literature to introduce deuterium atoms at the alpha positions to the carbonyl functions of keto-steroids were investigated6 without successfully incorporating deuterium into to the alpha-keto positions of 3,6-cholestandione (5). An alternative and practical procedure to promote this chemical transformation was developed. A solution of 3,6-cholestandione (5) in dioxane was added via a syringe to a solution of NaOD formed in situ. This reaction mixture was refluxed for 36 hours giving 2,2,4,4,5,7,7-d7-3,6-cholestandione (6)terium atoms were incorporated in the α position to both carbonyl functions and confirmed by GC-MS analysis.
Removal of the carbonyl functions of 6 under mild conditions was accomplished by treatment of this compound with tosylhydrazine in methanol under reflux for 3 hours. At that point, the formation of the tosylhydrazone intermediate (7) was isolated and was confirmed spectroscopically using NMR and MS. This intermediate-containing solution, without isolation or purification, was added to an excess of sodium borohydride7 and the reaction mixture was maintained under reflux for eight hours and worked up in the usual way to produce the new chemical biomarker 2,2,4,4,5,7,7-d7-cholestane (1) in yield of 73% after purification by flash chromatography. This is the first synthesis of (1), which we believe will find wide use as a biomarker due to the ease of preparation and high degree of deuteration.
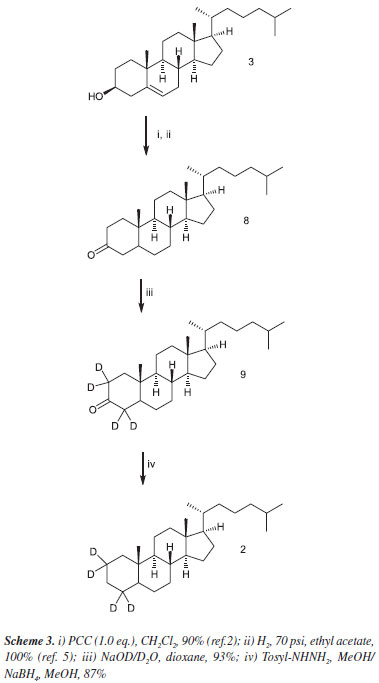
The same sequence of reactions was used to prepare the expensive, commercially available tetradeuterated cholestane (2) also in good yields (Scheme 3).
It is worth noting that the synthesis of deuterated cholestane structures 1 and 2 from cheap and easily available cholesterol (3) described herein can be used in environmental analysis laboratories to determinate the source of spills, which sometimes occur during petroleum extraction and transportation.8
CONCLUSIONS
Compound 1, a novel structure, presented on mass spectral analysis three specific peaks representing deuterated fragments from ring B (Scheme 4). This structural characteristic represents an additional advantage by eliminating the presence of spectral interferences which could be a problem with commercially available cholestane-d4 (2) currently used in quantitative studies of steranes from petroleum sources.2,3
For the next step in our analytical work1 related to samples of petroleum, we will investigate the use of 2,2,4,4,5,7,7-d7-cholestane (1), in quantification studies of sterane compounds in matrices from several areas of petroleum production in Brazil.
EXPERIMENTAL
General experimental procedures
Melting points were determined in a Melt-Temp apparatus. 1H and 13C NMR spectra were obtained in GEMINI-200MHZ spectrometer with Me4Si (δ = 0.00) as internal standard using as solvents CDCl3 or MeOD. The abbreviations used to describe multiplicity of the signals are s (singlet), d (doublet), dd (double dublet), t (triplet), m (multiplet). Chemical shifts were expressed as δ values (parts per million) utilizing tetramethylsilane as reference. Low resolution mass spectra were obtained in an Auto Specq, in the EI mode at 70 eV and high resolution spectra using a Varian MAT-CH7 instrument at 70 eV. High-resolution mass spectra were taken on an ATLAS MS-12, Consolidated 12-110 B, and FINNEGAN 400 mass spectrometers at 70 eV. All chemicals and solvents commercially available were purchased from Aldrich Co. (USA) or TEDIA-Brazil.
4-cholesten-3,6-dienone (4)
A continually stirred suspension of PCC (0.75 g, 3.50 mmol) in DCM (10 mL) was added to cholesterol (3) (0,193 g, 0.50 mmol) in DCM (10 mL). This reaction mixture was stirred under nitrogen at room temperature for 24 h after which it was filtered through a layer of silica gel and washed with diethyl ether (5 × 20 mL). The solvent was evaporated to give a crude brown product which was further purified by flash chromatography on silica gel column using as eluent a mixture of hexanes and ethyl acetate (4:1) affording the 4-cholesten-3,6-dienone (4) in a yield of 0.1865 g, (83%) as a pale yellow solid; mp: 116 ºC.
IR (KBr) 1712, 1688 cm-1.
1H NMR (200 MHz, CDCl3): δ = 6.17 (s, 1H), 0.85- 2.78 (m, 35H), 0.77 (s, 6H).
13C NMR (50 MHz, CDCl3): δ = 12.0 (C-18), 17.6 (C-19), 18.8 (C-21), 21.0 (C-11), 22.7 (C-26 and C-27), 23.9 (C-23), 24.1 (C-15), 28.1 (C-16 and C-25), 34.1 (C-2), 34.3 (C-l), 35.7 (C-9) 35.8 (C-20), 36.2 (C-22), 39.3 (C-10), 39.6 (C-24), 39.9 (C-12), 42.7 (C-13), 46.9 (C-7), 51.1 (C-8), 56.1 (C-17), 56.7 (C-14), 125.6 (C-4), 161.2 (C-5), 199.6 (C-3), 202.4 (C-6).
MS (EI, 70 eV): m/z (%) = 137 (100), 243 (90), 285 (33), 384 (75), 398 (50[M+.]).
HRMS (EI): m/z calcd. for C27H42O2: 398.3185; found: 398.3178.
Anal. Calcd. for C27H42O2: C, 81.35; H, 10.62. Found: C, 81.25; H, 10.59.
3,6-cholestandione (5)
For the hydrogenation reaction, 4-cholesten-3,6-dione (4, 0.207 g, 0.52 mmol) and 30 mg of Pd/C 10% was added 5 mL of ethyl acetate rature under 70 psi of hydrogen for 40 h. The suspension was filtered by vacuum filtration through silica gel, and the filtrate was evaporated giving the crude product, 3,6-cholestandione (5) in quantitative yield (0.208 g) as a white solid, which was used in the next step without purification; m.p. 169 ºC.
IR (KBr) 1712 cm-1.
1H NMR (200 MHz, CDCl3): δ = 0.69 (s, 3H), 0.8-2.6 (m, 41H), no signal between 5.5-7.0 ppm.
13C NMR (50 MHz, CDCl3): δ = 12.0 (C-18), 12.6 (C-19), 18.7 (C-21), 21.7 (C-11), 22.6 (C-26 e C-27), 23.8 (C-23), 24.0 (C-15), 28.0 (C-16 e C-25), 35.7 (C-20), 36.1 (C-22), 37.0 (C-8), 37.4 (C-1), 38.1 (C-2), 38.1 (C-4), 39.4 (C-24), 39.5 (C-12), 41.3 (C-10), 43.0 (C-13), 46.7 (C-7), 53.5 (C-9), 56.1 (C-14), 56.6 (C-17), 57.5 (C-5), 209.2 (C-3), 211.4 (C-6).
MS (EI, 70 eV): m/z (%) = 137 (20), 245 (100), 262 (15), 287 (60), 386 (25), 400 (100 [M+.]).
HRMS (EI): m/z calcd. for C27H44O2: 400.3341; found: 400.3337.
Anal. Calcd. for C27H44O2: C, 80.94; H, 11.07. Found: C, 80.81; H, 11.03.
3,6-cholestandione-2,2,4,4,5,7,7-d7 (6) and 3-cholestanone2,2,4,4-d4 (9)
A 25 mL round-bottomed flask equipped with a reflux condenser was charged with 3,6-cholestandione (5, 0.0259 g, 6.46 X 10-2 mmol) or (8, 0.025 g, 6.46 X 10-2 mmol) in 5 mL of dioxane. To this solution, 0.5 mL of 10% NaOD in D2O was added. The reaction mixture was refluxed for 36 hours and cooled to room temperature after which the solvent was removed under reduced pressure. The residue was dissolved in diethyl ether and washed successively with water. The organic phase was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude product was purified by flash chromatography using as eluent ethyl acetate and hexanes (1:9) giving 3,6-cholestandione-2,2,4,4,5,7,7-d7 (6) in a 91% yield (0,024 g) or 3-cholestanone-2,2,4,4-d4 (9), in a 93% yield, (0,023 g) both as white solids; m.p. 169 and 129 ºC respectively.
3,6-cholestandione-2,2,4,4,5,7,7-d7 (6)
IR (KBr) 2183 ν(C-D), 1715, 1691, cm-1.
1H NMR (200 MHz, CDCl3): δ = 0.70 (s, 3H), 0.8-2.3 (m, 34H), signal attenuation between 1.7-2.3 ppm
13C NMR (50 MHz, CDCl3): δ = 12.0 (C-18), 12.6 (C-19), 18.7 (C-21), 21.7 (C-11), 22.6 (C-26 and C-27), 23.8 (C-23), 24.0 (C-15), 28.0 (C-16 e C-25), 35.7 (C-20), 36.1 (C-22), 37.0 (C-8), 37.4 (C-1), 38.1 (C-2), 38.1 (C-4), 39.4 (C-24), 39.5 (C-12), 41.3 (C-10), 43.0 (C-13), 46.7 (C-7), 53.5 (C-9), 56.1 (C-14), 56.6 (C-17), 57.5 (C-5), 209.2 (C-3), 211.4 (C-6).
MS (EI, 70 eV): m/z (%) = 137 (40), 245 (100), 287 (60), 385 (25), 407 (100, [M+.]).
HRMS (EI): m/z calcd. for C27H37D7O2: 407.3781; found: 407.3786.
Anal. Calcd. for C27H37D7O22: C, 79.54; H, 12.61. Found C, 79.29; H, 12.59.
3-cholestanone-2,2,4,4-d4 (9)
IR (KBr) 2201 ν(C-D), 1715 cm-1.
1H NMR (200 MHz, CDCl3): δ = 0.7 (s, 3H), 0.8-2.0 (m, 39H), no signals between 2.0-2.5 ppm.
13C NMR (50 MHz, CDCl3): δ = 11.5 (C-19), 12.1 (C-18), 18.7 (C-21), 21.5 (C-11), 22.6 (C-26 e C-27), 23.9 (C-23), 24.3 (C-15), 28.1 (C-25), 28.3 (C-16), 29.1 (C-6), 31.8 (C-7) 35.5 (C-20), 35.7 (C-10), 35.9 (C-8), 36.2 (C-22) 38.3 (C-2), 38.6 (C-1), 39.6 (C-24), 40.0 (C-12), 42.7 (C-13), 44.8 (C-4), 46.8 (C-5), 53.9 (C-9), 56.4 (C-14 e C-17), 212.3 (C-3).
MS (EI, 70 eV): m/z (%) = 163 (15), 232 (100), 372 (25), 390 (20, [M+.]).
HRMS (EI): m/z calcd. for C27H42D4O: 390.3800; found: 390.3803.
2,2,4,4,5,7,7-d7-cholestane (1)
A 25 mL round-bottomed flask was charged with 3,6-cholestandione-2,2,4,4,5,7,7-d7 (6, 0.0701 g, 0.17 mmol) or (9, 3-cholestanone-2,2,4,4-d4, 0.0663 g, 0.17 mmol), tosylhydrazide (0.0900 g, 0.48 mmol) in 3.5 mL of methanol. The mixture is heated under gentle reflux for 3 hours and cooled to room temperature. To this solution 0.25 g was added (7.5 mmol) of sodium borohydride in small portions and the resulting mixture was heated under reflux for an additional 8 hours. The reaction mixture is cooled to room temperature and the solvent was removed under vacuum. The residue was dissolved in diethyl ether, transferred to a separatory funnel, and washed successively with water, a dilute aqueous solution of sodium carbonate, a solution of 2.0 M HCl, and water. The ethereal solution was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude product was purified by chromatography giving either 2,2,4,4,5,7,7-d7-cholestane (1) at a yield of 0.0476 g (73%) or 2,2,4,4-d4-cholestane (2), 87%, 0.056g both as a white solid; m.p. 79 ºC.
IR (KBr) 2183 ν(C-D), 1670 cm-1.
1H NMR (200 MHz, CDCl3): δ = 0.81-2.37 (m, 35H), 0.72 (s, 6H).
13C NMR (50 MHz, CDCl3): δ = 12.3 (C-18), 12.4 (C-19), 18.9 (C-21), 21.0 (C-11), 22.2 (C-2), 22.7 (C-26 e C27), 23.0 (C-23), 24.0 (C-15), 24.4 (C-3), 28.2 (C-25), 28.4 (C-16) 29.3 (C-6 e C-4), 32.4 (C-7), 35.7 (C-8) 36.0 (C-20), 36.4 (C-10 e C-22), 38.8 (C-1), 39.7 (C-24), 40.3 (C-12), 42.8 (C-13), 47.0 (C-5), 55.0 (C-9), 56.5 (C-17), 56.8 (C-14).
MS (EI, 70 eV): m/z (%) = 156 (30), 224 (100), 264 (15), 365 (50), 379 (10, [M+.]).
HRMS: m/z calcd. for C27H41D7: 379.4195; found: 379.4192.
Anal. Calcd. for C27H41D7: C, 85.40; H, 14.60. Found: C, 85.32; H, 14.57.
2,2,4,4-d4-cholestane (2)
IR (KBr) 2956, 2183 ν(C-D) cm-1.
1H NMR (200 MHz, CDCl3): δ = 0.7 (s, 3H), 0.8-1.9 (m, 41H).
13C NMR (50 MHz, CDCl3): δ = 12.1 (C-18), 12.3 (C-19), 18.7 (C-21), 20.9 (C-11), 22.3 (C-2), 22.6 (C-26 e C27), 23.9 (C-23), 24.3 (C-15), 26.9 (C-3), 28.1 (C-25), 28.3 (C-16) 29.1 (C-6), 29.2 (C-4), 32.3 (C-7), 35.6 (C-8) 35.9 (C-20), 36.3 (C-10 e C-22), 38.8 (C-1), 39.6 (C-24), 40.2 (C-12), 42.7 (C-13), 47.1 (C-5), 54.8 (C-9), 56.4 (C-17), 56.7 (C-14).
MS (EI, 70 eV): m/z (%) = 153 (25), 221 (100), 262, (50), 362 (50), 376 (10, [M+.].
HRMS: m/z calcd. for C27H44D4: 376.4007; found: 376.4009.
ACKNOWLEDGMENT
The authors gratefully acknowledge financial support from FAPERJ, CNPq and CENPES-PETROBRAS for the Master Science fellowship to M. G. de Miranda.
SUPPLEMENTARY MATERIAL
The 1H-NMR, 13C-NMR and mass spectra for compounds 1, 4, 5 and 6 are available at http://quimicanova.sbq.org.br, in PDF file, with free access.
REFERENCES
1. Lobão, M. M.; Cardoso, J. N.; Mello, M. R.; Brooks, P. W.; Lopes C. C.; Lopes, R. S. C.; Mar. Pollut. Bull. 2010,60,2263.
2. Peters, K. E.; Walters, C. C.; Moldowan, J. M.; The Biomarker Guide: Biomarkers and Isotopes in Petroleum Systems, 2nd ed., University Press: Cambridge, 2005.
3. Douglas, G. S.; Stout, S. A.; Uhler, A. D.; McCarthy, K. J.; Emsbo-Mattingly, S. D.; Advantages of quantitative chemical fingerprinting. Oil spill environmental forensics fingerprinting and source identification. Academic Press: New York, 2007.
4. Christensen, J. H.; Tomasi, G.; Hansen, A. B.; Environ. Sci. Technol. 2005,39,255.
5. Denance, M.; Guyot, M.; Samadi, M.; Steroids. 2006,71,599.
6. Gaertner, P.; Bica, K.; Felzmann, W.; Forsdahl, G.; Gmeiner, G.; Steroids. 2007,72,429; Numazawa, M.; Handa, W.; Chem. Pharm. Bull. 2006,54,554; Furuta, T.; Namekawa, T.; Shibasaki, H.; Kasuya, Y.; Steroids. 1999,64,805; Tokes, L.; Amos, B. A.; J. Org. Chem. 1972,37,4421; Tokes, L.; Jones, G.; Djerassi, C.; J. Am. Chem. Soc. 1968,90,5465; Nolin, B.; Jones, R. N.; Can. J. Chem. 1952,30,727; Fukushima, D. K.; Lieberman, S.; Praetz, B.; J. Am. Chem. Soc. 1950,72,5205.
7. Li, J. J.; Name Reactions, 2nd ed., Springer-Verlag, Berlin, 2003; Taber, D. F.; Joshi, P. V.; Comptes Rendus de l'Academie des Sciences, Serie IIc: Chimie 2001,4,557; Taylor, E. J.; Djerassi, C.; J. Am. Chem. Soc. 1976,98,2275; Caglioti, L.; Organic Syntheses, Collective VI. John Wiley & Sons, Inc.: New York, 1988; Borch, R. F.; Bernstein, M. D.; Durst, H. D.; J. Am. Chem. Soc. 1971,93,2897; Barton, D. H. R.; Ives, D. A. J.; Thomas, B. R. A.; J. Chem. Soc. 1955,2056.
8. Seifert, W. K.; Moldowan, J. M.; Geochim. Cosmochim. Acta 1978,42,77; Wardroper, A. M. K.; Brooks, P. W.; Geochim. Cosmochim. Acta 1977,41,499.
Recebido em 15/1/13; aceito em 18/4/13; publicado na web em 17/7/13
Supplementary Information
The supplementary material is available in pdf: [Supplementary material]
- 1. Lobão, M. M.; Cardoso, J. N.; Mello, M. R.; Brooks, P. W.; Lopes C. C.; Lopes, R. S. C.; Mar. Pollut. Bull 2010,60,2263.
- 2. Peters, K. E.; Walters, C. C.; Moldowan, J. M.; The Biomarker Guide: Biomarkers and Isotopes in Petroleum Systems, 2nd ed., University Press: Cambridge, 2005.
- 3. Douglas, G. S.; Stout, S. A.; Uhler, A. D.; McCarthy, K. J.; Emsbo-Mattingly, S. D.; Advantages of quantitative chemical fingerprinting. Oil spill environmental forensics fingerprinting and source identification Academic Press: New York, 2007.
- 4. Christensen, J. H.; Tomasi, G.; Hansen, A. B.; Environ. Sci. Technol 2005,39,255.
- 5. Denance, M.; Guyot, M.; Samadi, M.; Steroids. 2006,71,599.
- 6. Gaertner, P.; Bica, K.; Felzmann, W.; Forsdahl, G.; Gmeiner, G.; Steroids. 2007,72,429;
- Numazawa, M.; Handa, W.; Chem. Pharm. Bull 2006,54,554;
- Furuta, T.; Namekawa, T.; Shibasaki, H.; Kasuya, Y.; Steroids. 1999,64,805;
- Tokes, L.; Amos, B. A.; J. Org. Chem. 1972,37,4421;
- Tokes, L.; Jones, G.; Djerassi, C.; J. Am. Chem. Soc. 1968,90,5465;
- Nolin, B.; Jones, R. N.; Can. J. Chem 1952,30,727;
- Fukushima, D. K.; Lieberman, S.; Praetz, B.; J. Am. Chem. Soc. 1950,72,5205.
- 7. Li, J. J.; Name Reactions, 2nd ed., Springer-Verlag, Berlin, 2003;
- Taber, D. F.; Joshi, P. V.; Comptes Rendus de l'Academie des Sciences, Serie IIc: Chimie 2001,4,557;
- Taylor, E. J.; Djerassi, C.; J. Am. Chem. Soc. 1976,98,2275;
- Caglioti, L.; Organic Syntheses, Collective VI John Wiley & Sons, Inc.: New York, 1988;
- Borch, R. F.; Bernstein, M. D.; Durst, H. D.; J. Am. Chem. Soc. 1971,93,2897;
- Barton, D. H. R.; Ives, D. A. J.; Thomas, B. R. A.; J. Chem. Soc. 1955,2056.
- 8. Seifert, W. K.; Moldowan, J. M.; Geochim. Cosmochim. Acta 1978,42,77;
- Wardroper, A. M. K.; Brooks, P. W.; Geochim. Cosmochim. Acta 1977,41,499.
Publication Dates
-
Publication in this collection
04 Oct 2013 -
Date of issue
2013
History
-
Received
15 Jan 2013 -
Accepted
18 Apr 2013