Resumo
With the increase in life expectancy registered in the past few decades, the prevalence of various medical conditions related to aging has been observed, such as dementia and related neurodegenerative conditions. The number of patients afflicted with these conditions is expected to significantly increase in the coming years. The growing social impact of dementia underlines the need for research aimed at identifying and better understanding this type of condition. Among neurodegenerative diseases, amyloidogenic diseases, in particular Alzheimer's disease (AD), are currently the most common form of dementia. Over the years, several hypotheses have been raised regarding the etiology of AD, such as the cholinergic, glutamatergic, amyloid cascade, oligomeric, metallic and diabetes type 3 hypotheses. Unfortunately, no cure is yet available for this disease, only drugs that aid in controlling the symptoms. This review article conducts a comprehensive approach of the main etiological hypotheses of AD, as well as the treatment prospects associated with each hypothesis.
INTRODUÇÃO
As doenças neurodegenerativas são patologias caracterizadas pela destruição irreversível de certos neurônios, o que leva à perda progressiva e incapacitante de determinadas funções do sistema nervoso. Algumas delas são hoje consideradas as maiores causas de demência no mundo.11 Burns, A.; Iliffe, S.; British Med. J.2009 , 338, b158.
A doença de Alzheimer (DA) é uma patologia que, atualmente, representa a forma mais comum de demência em idosos. Em 2011, as estimativas indicavam 24 milhões de pessoas acometidas pela DA no mundo, e prevê-se que, até o ano de 2030, este número atinja 72 milhões.22 Reitz, C.; Brayne, C.; Mayeux, R.; Nat. Rev. Neurol.2011 , 7, 137. Não existem muitos dados a respeito da incidência da DA no Brasil; entretanto, estima-se que um milhão de pessoas sofram desta doença no país. Existe a necessidade de aprimorar esses dados, pois a DA aparenta ser subdiagnosticada no território brasileiro.33 Ferri, C. P.; Revista Brasileira de Psiquiatria2012 , 34, 371.,44 Herrera, E.; Caramelli, P.; Silveira, A. S.; Nitrini, R.;Alzheimer Dis. Assoc. Disord.2002 , 16, 103.
O primeiro estudo descrevendo esta doença foi publicado há mais de um século pelo psiquiatra e neuropatologista alemão Alois Alzheimer. Os sintomas relatados incluíam falhas na memória recente, paranóia e problemas comportamentais e de linguagem, assim como um cérebro atrófico e com sinais de deposições protéicas anômalas (observados em exames post-mortem ), as quais foram posteriormente denominadas placas senis e emaranhados neurofibrilares.55 Alzheimer, A.; Neurol. Central.1907 , 25, 1134.
De maneira geral, observa-se na DA o comprometimento das capacidades cognitivas dos pacientes, o que tende a tornar-se mais significativo com o passar dos anos. Comumente, a memória recente é a primeira a ser afetada, porém outras habilidades também são comprometidas com o progresso da doença, como, por exemplo, a capacidade de realizar cálculos e de usar objetos e ferramentas que fazem parte do cotidiano da pessoa acometida pela doença.66 Small, G. W.; Rabins, P. V.; Barry, P. P.; Buckholtz, N. S.; DeKosky, S. T.; Ferris, S. H.; Finkel, S. I.; Gwyther, L. P.; Khachaturian, Z. S.; Lebowitz, B. D.; McRae, T. D.; Morris, J. C.; Oakley, F.; Schneider, L. S.; Streim, J. E.; Sunderland, T.; Teri, L. A.; Tune, L. E.; JAMA, J. Am. Med. Assoc.1997 , 278, 1363.
Atualmente, podem ser diferenciadas duas formas de DA: a DA de início tardio (LOAD - do inglês, Late Onset Alzheimer's Disease ) e a DA familiar (FAD - do inglês, Familial Alzheimer's Disease ). A FAD é caracterizada por ser de surgimento prematuro, e, por isso também é chamada de DA de início precoce (do inglês, Early Onset Alzheimer's Disease ), ocorrendo antes dos 60 anos, com uma forte componente genética (transmissão mendeliana autossômica dominante), e representando de 1% a 6% de todos os casos de DA. Já a LOAD, a forma mais comum da doença, é caracterizada por ser de advento tardio (após os 60 anos) e possui um arquétipo muito complexo. Ambas as formas da doença são definidas pelas mesmas características patológicas, principalmente o decréscimo das funções cognitivas, afetando, sobretudo, a memória recente, a linguagem, a capacidade de julgamento, a atenção e as funções executivas.77 Bekris, L. M.; Yu, C. E.; Bird, T. D.; Tsuang, D. W.;Journal of Geriatric Psychiatry and Neurology2010 , 23, 213.
Os dados neuropatológicos mais relevantes em pacientes de DA são a presença de atrofia cortical difusa, degeneração neurovascular, perdas neuronais e sinápticas envolvendo vários sistemas de neurotransmissão, presença de placas senis extracelulares compostas de agregados filamentosos da proteína β-amiloide (Aβ) e massas neurofibrilares intracelulares, formadas principalmente pela proteína tau.88 Serrano-Pozo, A.; Frosch, M. P.; Masliah, E.; Hyman, B. T.;Cold Spring Harbor Perspect. Biol.2011 , 1, a006189. Apesar de ser possível a presença destas alterações no cérebro de idosos sadios, os sintomas não são observados conjuntamente e nem com a mesma intensidade do que em pacientes acometidos pela DA.99 Smith, M. A. C.; Revista Brasileira de Psiquiatria1999 , 21, 03. O falecimento costuma ocorrer entre 6 e 12 anos após o início da doença, normalmente por uma complicação da imobilidade ou por embolia pulmonar e pneumonia.1010 Hardman, J. G.; Limbird, L. E.; Gilman, A. G.; Goodman, L. S.; Gilman, A.; Goodman & Gilman's the pharmacological basis of therapeutics, McGraw-Hill: New York, 1996.
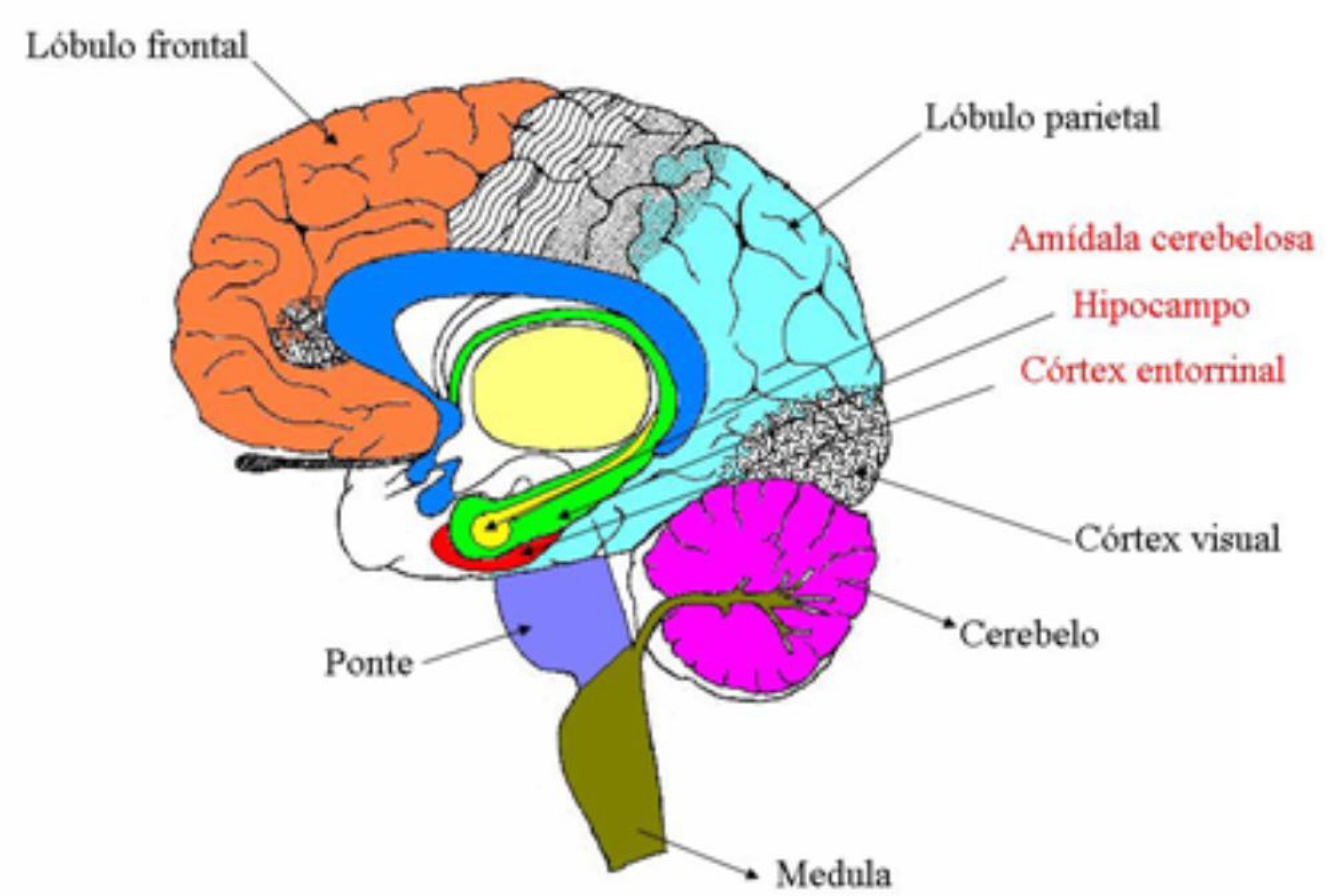
As placas e massas citadas anteriormente, presentes no cérebro de pacientes acometidos por DA estão localizadas, sobretudo, nas amídalas cerebelosas, no hipocampo e no córtex entorrinal do lóbulo temporal, enquanto as porções parietais e frontais do córtex associativo são menos afetadas.88 Serrano-Pozo, A.; Frosch, M. P.; Masliah, E.; Hyman, B. T.;Cold Spring Harbor Perspect. Biol.2011 , 1, a006189. A Figura 1 mostra um esquema das diferentes regiões do cérebro, com as áreas mais afetadas pela DA em destaque.
Diversos outros sinais bioquímicos também são observados na DA, como, por exemplo, estresse oxidativo difundido no cérebro, neuroinflamação, desregulação de cálcio, deficiência e distribuição alterada das mitocôndrias, oligomerização do peptídeo Aβ, toxicidade sináptica e problemas na homeostase metálica, os quais serão discutidos adiante.
BASES MOLECULARES DA DOENÇA DE ALZHEIMER
Diferentes hipóteses a respeito das bases moleculares da DA têm sido levantadas, modificando-se gradualmente devido aos diversos avanços tecnológicos ocorridos ao longo do tempo. A teoria mais antiga é representada pela hipótese colinérgica, postulada no início da década de 80. Já na metade dessa década, há o surgimento da hipótese glutamatérgica. A hipótese da "cascata amiloide" foi primeiramente proposta em 1992. Outras conjecturas, como as hipóteses oligomérica e metálica, podem ser consideradas extensões da hipótese amiloide e começaram a tomar maiores proporções durante a década de 90. A hipótese mais recentemente postulada correlaciona a DA com o diabetes, originando o termo "diabetes de tipo 3". Cada uma destas hipóteses será abordada em maiores detalhes nas próximas seções.
A hipótese colinérgica
A hipótese mais antiga sobre a DA foi introduzida no início da década de 80, e descrita como a hipótese colinérgica na disfunção amnésica do idoso.1111 Bartus, R. T.; Dean, R. L.; Beer, B.; Lippa, A. S.;Science1982 , 217, 408.,1212 Coyle, J. T.; Price, D. L.; DeLong, M. R.; Science1983 , 219, 1184. A importância da função colinérgica nos processos de aprendizagem e memória é conhecida desde o início da década de 70,1313 Deutsch, J. A.; Science1971 , 174, 788. e as pesquisas a respeito da importância do sistema colinérgico na DA demonstraram diversas características, como a diminuição na concentração da colina acetiltransferase (ChAT), enzima responsável pela síntese da acetilcolina (ACh), no córtex e no hipocampo, assim como uma redução variável dos neurônios colinérgicos localizados no núcleo basal de Meynert.1414 Davies, P.; Maloney, A. J.; Lancet1976 , 2, 1403.,1515 Kása, P.; Rakonczay, Z.; Gulya, K.; Prog. Neurobiol.1997 , 52, 511. Uma observação pertinente foi a da associação positiva entre essas duas depleções e o grau de severidade do déficit cognitivo do paciente em vida.1515 Kása, P.; Rakonczay, Z.; Gulya, K.; Prog. Neurobiol.1997 , 52, 511.,1616 Wilcock, G. K.; Esiri, M. M.; Bowen, D. M.; Smith, C. C.; J. Neurol. Sci.1982 , 57, 407. Estudos subsequentes demonstraram que a administração de substâncias colinomiméticas reduzia as dificuldades mnemônicas apresentadas por pessoas acometidas pela doença.1717 Drachman, D. A.; Sahakian, B. J.; Arch. Neurol.1980 , 37, 674.,1818 Christie, J. E.; Shering, A.; Ferguson, J.; Glen, A. I.; Br. J. Psychiatry1981 , 138, 46.
Estudos realizados em mamíferos também investigaram os efeitos dos inibidores irreversíveis da acetilcolinesterase (AChE, enzima responsável por catalisar a hidrólise da acetilcolina restante no espaço sináptico) sobre a aprendizagem, assim como a relação entre os níveis cerebrais de AChE e o desempenho de cobaias em testes de reconhecimento espacial.1919 Bennett, B. M.; Reynolds, J. N.; Prusky, G. T.; Douglas, R. M.; Sutherland, R. J.; Thatcher, G. R.; Neuropsychopharmacology2007 , 32, 505.,2020 Muthuraju, S.; Maiti, P.; Solanki, P.; Sharma, A. K.; Amitabh; Singh, S. B.; Prasad, D.; Ilavazhagan, G.; Behav. Brain Res.2009 , 203, 1. Estas pesquisas apontaram a melhora da aprendizagem devido à ativação do sistema colinérgico. A redução do desempenho de aprendizagem e memória em diferentes modelos animais expostos à administração de antagonistas muscarínicos,i.e . escopolamina, também corrobora estes dados.2121 Berger-Sweeney, J.; Arnold, A.; Gabeau, D.; Mills, J.;Behav. Neurosci.1995 , 109, 859. Estudos farmacológicos em seres humanos têm mostrado que este tipo de substância impede a formação de novas memórias sem influenciar a reaquisição de eventos do passado remoto.2222 Hasselmo, M. E.; Curr. Opin. Neurobiol.2006 , 16, 710. Pelo contrário, as substâncias com ação colinomimética promovem a aquisição de novos traços de memória, tanto em seres humanos quanto em modelos animais.2323 Buccafusco, J. J.; Letchworth, S. R.; Bencherif, M.; Lippiello, P. M.; Trends Pharmacol. Sci.2005 , 26, 352. Isto demonstra a importância da ativação dos receptores colinérgicos no processo de fixação da memória. Recentemente, tem sido demonstrado que o antagonismo dos receptores nicotínicos e muscarínicos causa deterioração cognitiva, indicando que este processo pode ser governado pelo funcionamento de ambos os receptores, os quais seriam ativados na modulação dos processos mnésicos, interagindo reciprocamente.2424 Green, A.; Ellis, K. A.; Ellis, J.; Bartholomeusz, C. F.; Ilic, S.; Croft, R. J.; Phan, K. L.; Nathan, P. J.; Pharmacol. Biochem. Behav.2005 , 81, 575.
A hipótese da disfunção glutamatérgica
A hipótese glutamatérgica, conhecida também como "excito-tóxica", da DA também emergiu na década de 80.2525 Greenamyre, J. T.; Maragos, W. F.; Albin, R. L.; Penney, J. B.; Young, A. B.; Prog. Neuro-Psychopharmacol. Biol. Psychiatry1988 , 12, 421. O glutamato, o principal neurotransmissor excitatório do sistema nervoso central, tem sua atividade mediada por receptores de 3 tipos: de N-metil-d-Aspartato (NMDA), do ácido α-amino-3-hidroxi-5-metil-4-isoxazolepropiônico (AMPA) e de cianato. A hipótese glutamatérgica prevê que, em condições especificas, tais como, por exemplo, a alteração do metabolismo energético celular, ocorre uma excessiva ativação de receptores de NMDA, podendo alterar a homeostase de cálcio, levando a um aumento das concentrações intracelulares deste metal capaz de iniciar o processo de apoptose (degeneração e morte) neuronal.2626 Danysz, W.; Parsons, C. G.; Mobius, H. J.; Stoffler, A.; Quack, G.;Neurotox. Res.2000 , 2, 85.,2727 Parsons, C. G.; Stöffler, A.; Danysz, W.;Neuropharmacology2007 , 53, 699. Os receptores de NMDA apresentam uma estrutura complexa, com diferentes sítios de ligação para o glutamato e diversos moduladores. A ativação fisiológica destes receptores gera, em nível neuronal, um fluxo de íons Ca2+. Acredita-se que o glutamato liberado ative inicialmente os receptores de AMPA e de cianato, que, apesar da baixa afinidade, promoveriam a rápida despolarização da célula por meio da entrada de íons Na+ e Ca2+. A entrada desses íons provocaria uma despolarização parcial da membrana plasmática, removendo o bloqueio exercido pelos íons Mg2+ dentro do canal do receptor de NMDA. Assim, o glutamato poderia ligar-se a uma subunidade deste receptor promovendo, em associação com a glicina ligada a outra subunidade, a entrada de mais Ca2+ e Na+ no neurônio, contribuindo para a sua excitabilidade.2828 Sucher, N. J.; Awobuluyi, M.; Choi, Y. B.; Lipton, S. A.;Trends Pharmacol. Sci.1996 , 17, 348.,2929 Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S. F.;Pharmacol. Rev.1999 , 51, 7. Postula-se, portanto, que essa "excito-toxicidade" glutamato-dependente possa constituir um dos mecanismos patogênicos necessários para que o processo neurodegenerativo seja mantido e amplificado.3030 Greenamyre, J. T.; Young, A. B.; Neurobiol. Aging1989 , 10, 593.
A hipótese da cascata amiloide
Desde a descoberta da DA, é reconhecido que os sintomas da doença podem ser associados ao desenvolvimento de inúmeras lesões filamentosas intraneuronais e extracelulares no córtex límbico, assim como no córtex cerebral. Agregados anormais de fibras citoplasmáticas ocorrem tanto nos corpos celulares neuronais, envolvendo os emaranhados neurofibrilares, quanto nos axônios e dendritos. Estes sintomas são chamados coletivamente de neurites distróficas. Juntamente com a presença das neurites distróficas, há também outro importante sinal histopatológico na DA: a difundida presença de placas e agregados, formados principalmente pelo peptídeo Aβ, na porção extracelular do tecido cerebral.3131 Nie, Q.; Du, X. G.; Geng, M. Y.; Acta Pharmacol. Sin.2011 , 32, 545.
32 Jenkins, E. C.; Devine-Gage, E. A.; Robakis, N. K.; Yao, X. L.; Brown, W. T.; Houck, G. E.; Wolfe, G.; Ramakrishna, N.; Silverman, W. P.; Wisniewski, H. M.; Biochem. Biophys. Res. Commun.1988 , 151, 1.-3333 Selkoe, D. J.; J. Biol. Chem.1996 , 271, 18295. A Figura 2 apresenta, esquematicamente, as diferenças entre um neurônio saudável e um neurônio característico de um paciente com DA.
Diferenças esquemáticas entre um neurônio saudável (A) e um neurônio de um paciente com DA (B)
Embora deposições de origem amiloide (peptídeo Aβ) possam ser também detectadas em pequenas quantidades em cérebros de idosos sadios, a produção deste tipo de peptídeo é considerada central na patologia da DA. O trabalho que pela primeira vez propôs a sequência de eventos denominada "hipótese da cascata amiloide" foi publicado no início da década de 90 e postula que o peptídeo Aβ e/ou os produtos de clivagem da sua proteína precursora, uma glicoproteína integral denominada proteína precursora amiloide (APP), são neurotóxicos e podem levar à formação das placas senis, resultando em morte celular.3434 Hardy, J. A.; Higgins, G. A.; Science1992 , 256, 184. Neste contexto, tem se associado o fato de que pacientes com trissomia do cromossomo 21 (síndrome de Down) apresentam depósitos de Aβ no final da infância ou no início da idade adulta e, posteriormente, desenvolvem as características neuropatológicas clássicas da DA, quando atingem por volta de quarenta anos, devido à localização do gene que codifica a APP, justamente no cromossomo 21.3535 Giaccone, G.; Tagliavini, F.; Linoli, G.; Bouras, C.; Frigerio, L.; Frangione, B.; Bugiani, O.; Neurosci. Lett.1989 , 97, 232.,3636 Iwatsubo, T.; Mann, D. M.; Odaka, A.; Suzuki, N.; Ihara, Y.;Ann. Neurol.1995 , 37, 294.Esta constatação conduziu a uma procura específica por famílias com autossomia dominante de DA que tivessem ligação genética com o cromossomo 21, resultando assim na identificação de seis diferentes mutações, sendo cinco associadas à FAD,3737 Goate, A.; Chartier-Harlin, M. C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.;Nature1991 , 349, 704.
38 Chartier-Harlin, M. C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.;Nature1991 , 353, 844.
39 , J.; Farlow, M.; Ghetti, B.; Benson, M. D.;Science1991 , 254, 97.
40 Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L.; Nat. Genet.1992 , 1, 345.-4141 Hendriks, L.; van Duijn, C. M.; Cras, P.; Cruts, M.; Van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M. G.; Antonarakis, S. E.; Martin, J. J.;Nat. Genet.1992 , 1, 218. e uma associada à síndrome da hemorragia cerebral hereditária com amiloidose do tipo holandês.4242 Levy, E.; Carman, M. D.; Fernandez-Madrid, I. J.; Power, M. D.; Lieberburg, I.; van Duinen, S. G.; Bots, G. T.; Luyendijk, W.; Frangione, B.;Science1990 , 248, 1124.
A formação do peptídeo Aβ, composto de 39-42 aminoácidos, é o resultado da digestão da APP, que aparenta ter função fisiológica fundamental com relação aos fenômenos de neuroplasticidade.4343 Haass, C.; Schlossmacher, M. G.; Hung, A. Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B. L.; Lieberburg, I.; Koo, E. H.; Schenk, D.; Teplow, D. B.; Nature1992 , 359, 322.,4444 Mattson, M. P.; Physiol. Rev.1997 , 77, 1081. Observou-se que diversos fragmentos com funções fisiológicas e fisiopatológicas são gerados a partir dessa proteína precursora.3333 Selkoe, D. J.; J. Biol. Chem.1996 , 271, 18295.,4444 Mattson, M. P.; Physiol. Rev.1997 , 77, 1081.
45 Nitsch, R. M.; Farber, S. A.; Growdon, J. H.; Wurtman, R. J.;Proc. Natl. Acad. Sci. U. S. A.1993 , 90, 5191.-4646 Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A. Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D. J.;Nature1992 , 360, 672. Duas secretases, a γ-secretase e a β-secretase, clivam a APP em diferentes lugares, originando fragmentos de Aβ de diferentes tamanhos, com 40 e 42 resíduos, respectivamente: Aβ1-40 e Aβ1-42. Apesar do primeiro fragmento ser o mais comum, atualmente considera-se que o segundo, mais hidrofóbico, possui um maior potencial amiloidogênico, embora ambos sejam capazes de se agregar e originar protofibrilas, fibrilas e, por fim, placas insolúveis.4747 Soreghan, B.; Kosmoski, J.; Glabe, C.; J. Biol. Chem.1994 , 269, 28551.
Diferentes estudos têm apontado para a correlação entre mutações de APP e γ-secretase e algumas formas raras de DA familiar.3737 Goate, A.; Chartier-Harlin, M. C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.;Nature1991 , 349, 704.,3838 Chartier-Harlin, M. C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.;Nature1991 , 353, 844.,4040 Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L.; Nat. Genet.1992 , 1, 345.,4141 Hendriks, L.; van Duijn, C. M.; Cras, P.; Cruts, M.; Van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M. G.; Antonarakis, S. E.; Martin, J. J.;Nat. Genet.1992 , 1, 218.,4646 Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A. Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D. J.;Nature1992 , 360, 672.,4848 Finckh, U.; Kuschel, C.; Anagnostouli, M.; Patsouris, E.; Pantes, G. V.; Gatzonis, S.; Kapaki, E.; Davaki, P.; Lamszus, K.; Stavrou, D.; Gal, A.;Neurogenetics2005 , 6, 85.
49 Renbaum, P.; Levy-Lahad, E.; Cell. Mol. Life Sci.1998 , 54, 910.
50 Tanzi, R. E.; Kovacs, D. M.; Kim, T. W.; Moir, R. D.; Guenette, S. Y.; Wasco, W.; Neurobiol Dis.1996 , 3, 159.
51 Tanzi, R.; Gaston, S.; Bush, A.; Romano, D.; Pettingell, W.; Peppercorn, J.; Paradis, M.; Gurubhagavatula, S.; Jenkins, B.; Wasco, W.;Genetica1993 , 91, 255.
52 Wasco, W.; Peppercorn, J.; Tanzi, R. E.; Ann. N. Y. Acad. Sci.1993 , 695, 203.
53 Taylor, J. E.; Tinklenberg, J. R.; Eng, L. F.; Yesavage, J. A.; Vinogradov, S.; Davies, H. G.; Gonzalez-DeWhitt, P. A.; Frossard, P. M.;Mol. Biol. Med.1988 , 5, 167.-5454 Haass, C.; Hung, A. Y.; Selkoe, D. J.; Teplow, D. B.; J. Biol. Chem.1994 , 269, 17741. Diversos modelos animais da doença corroboram este achado, pois apresentam uma superexpressão de β-APP e presenilina, proteína enzimática de membrana responsável pela regulação das γ-secretases.5555 Spires, T. L.; Hyman, B. T.; NeuroRx2005 , 2, 423.
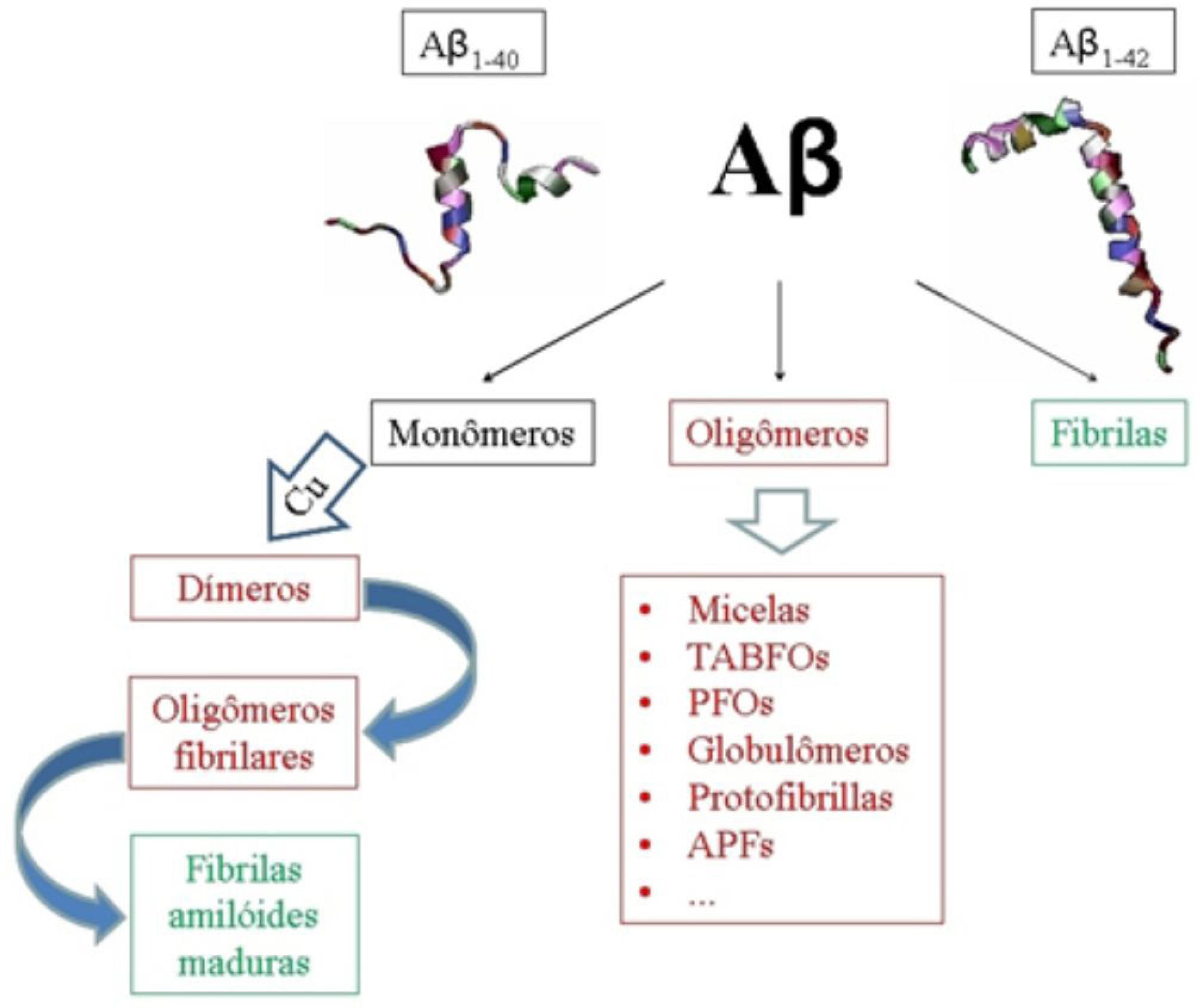
Diferentes formas do peptídeo Aβ foram identificadas, com funções e propriedades específicas.5656 Saido, T. C.; Iwatsubo, T.; Mann, D. M.; Shimada, H.; Ihara, Y.; Kawashima, S.; Neuron1995 , 14, 457.,5757 He, W.; Barrow, C. J.; Biochemistry1999 , 38, 10871. Aβ solúvel, por exemplo, foi identificado na metade da década de 90 a partir de frações solúveis retiradas do córtex cerebral de pacientes com DA, aparentando ser a primeira forma de acúmulo do peptídeo.5858 Tabaton, M.; Nunzi, M. G.; Xue, R.; Usiak, M.; Autiliogambetti, L.; Gambetti, P.; Biochem. Biophys. Res. Commun.1994 , 200, 1598. O peptídeo Aβ pode ser encontrado na forma de monômeros e em estruturas compostas por dímeros e trímeros, formando as denominadas arquiteturas "monoméricas" ou "oligoméricas", enquanto as "protofibrilas" representam as estruturas de ordem intermediária entre os agregados mencionados e as fibrilas presentes nas placas senis.5959 Gandy, S.; J. Clin. Invest.2005 , 115, 1121. O termo oligômero caracteriza um amplo grupo de agregados não-fibrilares. Entre as espécies identificadas como oligômeros incluem-se, como mostrado esquematicamente na Figura 3, agregados desordenados, micelas, protofibrilas, agregados pré-fibrilares, oligômeros fibrilares tóxicos de Aβ (TABFOs - do inglês, Toxic Amyloid-Beta Fibrillar Oligomers ), ligantes amiloides difusíveis, oligômeros pré-fibrilares (PFOs - do inglês, Prefibrillar Oligomers ), globulômeros e protofibrilas anulares (APFs - do inglês, Annular Protofibrils ), que agem como canais de íons Ca2+, afetando a homeostase celular e proporcionando uma explicação biofísica direta para o mecanismo subjacente a certos sintomas da DA.6060 Hane, F.; Leonenko, Z.; Biomolecules2014 , 4, 101.
Verificou-se recentemente que a estrutura em grampo do Aβ pode rearranjar-se em diferentes conformações, o que explicaria a variedade de espécies oligoméricas observadas.6161 Gu, L.; Liu, C.; Guo, Z.; J. Biol. Chem.2013 , 288, 18673. As estruturas dos oligômeros e a formação das fibrilas são consideravelmente dependentes do ambiente celular. Por exemplo, protofibrilas são instáveis em solução, mas podem ser estabilizadas por superfícies. Superfícies hidrofóbicas promovem a agregação de estruturas amorfas, e superfícies carregadas promovem a formação fibrilar.6262 Moores, B.; Drolle, E.; Attwood, S. J.; Simons, J.; Leonenko, Z.;PLoS One2011 , 6, e25954.
63 Lee, G.; Lee, W.; Lee, H.; Woo Lee, S.; Sung Yoon, D.; Eom, K.; Kwon, T.; Appl. Phys. Lett.2012 , 101, 043703.-6464 Burke, K. A.; Yates, E. A.; Legleiter, J.; Front. Neurol.2013 , 4, 17. A agregação de monômeros de Aβ é passível de ocorrer por meio de diferentes caminhos. Inicialmente, pode haver a dimerização do peptídeo formando oligômeros fibrilares que, em seguida, polimerizam-se para formar fibrilas amiloides maduras.6060 Hane, F.; Leonenko, Z.; Biomolecules2014 , 4, 101.,6565 Hane, F.; Tran, G.; Attwood, S. J.; Leonenko, Z.; PLoS One2013 , 8, e59005. O mesmo dímero pode formar oligômeros pré-fibrilares, gerando em seguida protofibrilas que podem sofrer uma mudança conformacional em bloco para formar fibrilas amiloides.6666 Lee, J.; Culyba, E. K.; Powers, E. T.; Kelly, J. W.; Nat. Chem. Biol.2011 , 7, 602.,6767 Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C. B.; Condron, M. M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D. B.; Younkin, S. G.; Näslund, J.; Lannfelt, L.; Nat. Neurosci.2001 , 4, 887. Biometais, como o cobre, podem mediar a dimerização dos monômeros de Aβ, formando pequenos oligômeros de Aβ-Cu e, eventualmente, levando à formação de agregados maiores.6060 Hane, F.; Leonenko, Z.; Biomolecules2014 , 4, 101.,6565 Hane, F.; Tran, G.; Attwood, S. J.; Leonenko, Z.; PLoS One2013 , 8, e59005.
Os oligômeros e as protofibrilas aparentam ser os estados de aglomeração de maior toxicidade.6868 Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T. M.; Milton, S. C.; Hall, J. E.; Glabe, C. G.; J. Biol. Chem.2004 , 279, 46363.,6969 Klein, W. L.; Krafft, G. A.; Finch, C. E.; Trends Neurosci.2001 , 24, 219. Estudos in vitrodemonstraram que os oligômeros de Aβ exercem uma forte ação inibitória sobre a potenciação de longa duração, um fenômeno de melhoria duradoura na transmissão do sinal entre dois neurônios, que contribui para a plasticidade sináptica.7070 Puzzo, D.; Vitolo, O.; Trinchese, F.; Jacob, J. P.; Palmeri, A.; Arancio, O.; J. Neurosci.2005 , 25, 6887.
71 Selkoe, D. J.; Science2002 , 298, 789.-7272 Walsh, D. M.; Klyubin, I.; Fadeeva, J. V.; Cullen, W. K.; Anwyl, R.; Wolfe, M. S.; Rowan, M. J.; Selkoe, D. J.; Nature2002 , 416, 535.
O papel da proteína tau: possivelmente uma consequência
Análises imunocitoquímicas e bioquímicas dos emaranhados neurofibrilares intraneuronais levaram à conclusão de que outra proteína, denominada proteína tau, associada aos microtúbulos, é a principal ou, mais provavelmente, a única subunidade dos filamentos helicoidais emparelhados encontrados nesses emaranhados, assim como em muitas das neurites distróficas observadas no córtex de pacientes com DA.7373 Wishik, C. M.; Novak, M.; Edwards, P. C.; Klug, A.; Tichelaar, W.; Crowther, R. A.; Proc. Natl. Acad. Sci. U. S. A.1988 , 85, 4884. Diversos estudos demonstraram que esta proteína, normalmente solúvel, encontra-se hiperfosforilada em quadros de DA. Isto acaba tornando-a um polímero insolúvel filamentoso, o que parece desregular a cascata citoplasmática de fosforilações e desfosforilações.3333 Selkoe, D. J.; J. Biol. Chem.1996 , 271, 18295.,7474 Selkoe, D.; Mandelkow, E.; Holtzman, D.; Cold Spring Harbor Perspect. Med.2012 , 2, a011460. Diversos estudos indicam que o acúmulo de Aβ pode ser o evento ativador da hiperfosforilação da proteína tau, porém os fatores que desencadeiam este desequilíbrio ainda não são bem entendidos.3333 Selkoe, D. J.; J. Biol. Chem.1996 , 271, 18295.,7575 Zheng, W. H.; Bastianetto, S.; Mennicken, F.; Ma, W.; Kar, S.;Neuroscience2002 , 115, 201.
76 Lloret, A.; Badia, M. C.; Giraldo, E.; Ermak, G.; Alonso, M. D.; Pallardó, F. V.; Davies, K. J.; Viña, J.; J. Alzheimer's Dis.2011 , 27, 701.
77 Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B. A.;Neuron1995 , 14, 879.
78 Stancu, I. C.; Vasconcelos, B.; Terwel, D.; Dewachter, I.;Mol. Neurodegener.2014 , 9, 51.
79 Hu, X.; Li, X.; Zhao, M.; Gottesdiener, A.; Luo, W.; Paul, S.;Mol. Neurodegener.2014 , 9, 52.-8080 Annamalai, B.; Won, J. S.; Choi, S.; Singh, I.; Singh, A. K.;Biochem. Biophys. Res. Commun.2015 , 458, 214. Emaranhados neurofibrilares que contêm proteínas tau hiperfosforiladas são encontrados também em outras doenças neurológicas, sugerindo fortemente que estas alterações do citoesqueleto possam ser uma resposta secundária, embora de vital importância, a diversas lesões cerebrais.8181 Blennow, K.; de Leon, M. J.; Zetterberg, H.; Lancet2006 , 368, 387.
82 Weiner, M. W.; Veitch, D. P.; Aisen, P. S.; Beckett, L. A.; Cairns, N. J.; Green, R. C.; Harvey, D.; Jack, C. R.; Jagust, W.; Liu, E.; Morris, J. C.; Petersen, R. C.; Saykin, A. J.; Schmidt, M. E.; Shaw, L.; Siuciak, J. A.; Soares, H.; Toga, A. W.; Trojanowski, J. Q.; Initiative, A. s. D. N.;Alzheimer's Dementia2012 , 8, S1.-8383 Mudher, A.; Lovestone, S.; Trends Neurosci.2002 , 25, 22.
A hipótese oligomérica
A despeito do fato da agregação do peptídeo Aβ ser uma das características mais importantes para a caracterização da patogênese da DA, a função das placas extracelulares compostas por esta proteína ainda não está totalmente esclarecida. Existem evidências de que Aβ1-42 seja a forma mais tóxica do peptídeo.8484 Urbanc, B.; Betnel, M.; Cruz, L.; Li, H.; Fradinger, E. A.; Monien, B. H.; Bitan, G.; J. Mol. Biol.2011 , 410, 316. Na presença de Aβ1-42, os neurônios do córtex e do hipocampo também sofrem modificações, resultando na indução de déficits cognitivos e mnemônicos, mesmo sem ocorrer a morte neuronal.8585 Danysz, W.; Parsons, C. G.; Br. J. Pharmacol.2012 , 167, 324. O acúmulo progressivo do peptídeo, verificado experimentalmente por meio da administração repetida de Aβ em ratos, e a série de eventos que levam à formação das placas são, portanto, considerados importantes mecanismos nas fases iniciais de perda da memória.8686 Yamada, M.; Chiba, T.; Sasabe, J.; Nawa, M.; Tajima, H.; Niikura, T.; Terashita, K.; Aiso, S.; Kita, Y.; Matsuoka, M.; Nishimoto, I.;Behav. Brain Res.2005 , 164, 139.
Na década de 90, verificou-se que o peptídeo Aβ, além de formar fibrilas, também possui a capacidade de se agrupar em oligômeros solúveis.8787 Beyreuther, K.; Multhaup, G.; Masters, C. L.; Ciba Found. Symp.1996 , 199, 119. Um estudo publicado em 1998 demonstrou que o Aβ, quando forçado a permanecer em sua forma oligomérica, danifica imediatamente as sinapses neuronais, levando à morte celular, embora o mesmo não ocorra com as fibrilas de Aβ.8888 Lambert, M. P.; Barlow, A. K.; Chromy, B. A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T. E.; Rozovsky, I.; Trommer, B.; Viola, K. L.; Wals, P.; Zhang, C.; Finch, C. E.; Krafft, G. A.; Klein, W. L.; Proc. Natl. Acad. Sci. U. S. A.1998 , 95, 6448.Esse estudo e outros mais recentes demonstraram que os oligômeros induzem rapidamente a falha de plasticidade sináptica.7272 Walsh, D. M.; Klyubin, I.; Fadeeva, J. V.; Cullen, W. K.; Anwyl, R.; Wolfe, M. S.; Rowan, M. J.; Selkoe, D. J.; Nature2002 , 416, 535.,8989 Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R. P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J. M.; Grüning, B. A.; Wang, Q.; Schmidt, M. R.; Lurz, R.; Anwyl, R.; Schnoegl, S.; Fändrich, M.; Frank, R. F.; Reif, B.; Günther, S.; Walsh, D. M.; Wanker, E. E.; Nat. Chem. Biol.2012 , 8, 93. Ao longo dos anos, evidências têm sido coletadas, tanto a partir de estudos in vitro quanto com modelos animais, corroborando a hipótese de que a forma oligomérica é a que melhor explica a neurotoxicidade de Aβ.9090 Knobloch, M.; Farinelli, M.; Konietzko, U.; Nitsch, R. M.; Mansuy, I. M.; J. Neurosci.2007 , 27, 7648.
91 Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O.; J. Neurosci.2008 , 28, 14537.
92 Koffie, R. M.; Hyman, B. T.; Spires-Jones, T. L.; Mol. Neurodegener.2011 , 6, 63.
93 Ferreira, S. T.; Klein, W. L.; Neurobiol. Learn. Mem.2011 , 96, 529.
94 Selkoe, D. J.; Behav. Brain Res.2008 , 192, 106.-9595 Lue, L. F.; Kuo, Y. M.; Roher, A. E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J. H.; Rydel, R. E.; Rogers, J.; Am. J. Pathol.1999 , 155, 853.
Foram obtidas, ao longo das últimas duas décadas, provas circunstanciais sobre a correlação direta entre o acúmulo de Aβ e a agregação da proteína tau, que representaria o último estágio da patogênese da doença.9696 Lacor, P. N.; Buniel, M. C.; Chang, L.; Fernandez, S. J.; Gong, Y.; Viola, K. L.; Lambert, M. P.; Velasco, P. T.; Bigio, E. H.; Finch, C. E.; Krafft, G. A.; Klein, W. L.; J. Neurosci.2004 , 24, 10191. Os oligômeros de Aβ também têm sido responsabilizados diretamente pela formação de oligômeros de tau, em células neuronais em cultura.9797 Selkoe, D. J.; Cold Spring Harbor Perspect. Biol.2011 , 3, a004457. O mecanismo de danificação das sinapses por estes oligômeros, levando à morte de apenas alguns neurônios ou regiões neuronais específicas,9393 Ferreira, S. T.; Klein, W. L.; Neurobiol. Learn. Mem.2011 , 96, 529. ainda é desconhecido, assim como as dinâmicas da autopropagação das fibrilas e dos oligômeros.9898 Klein, W. L.; J. Alzheimer's Dis.2013 , 33, S49. Assim, a questão fundamental nos estudos que abordam essa hipótese é a verificação do mecanismo a partir do qual os oligômeros seriam capazes de alterar a composição e a morfologia das sinapses, causando a rápida perda de plasticidade.9999 Schnabel, J.; Nature2011 , 475, S12.
Correlação entre a hipótese amiloide e a colinérgica
O mecanismo de regulação do processamento da APP pelo caminho amiloidogênico, conforme visto anteriormente, ainda é objeto de investigação. Diferentes estudos sugerem que os inputs colinérgicos estão presentes neste processo.100100 Parikh, V.; Bernard, C. S.; Naughton, S. X.; Yegla, B.;Behav. Brain Res.2014 , 274, 30. A formação dos agregados extracelulares aparenta induzir uma resposta inflamatória capaz de danificar as células do sistema colinérgico.101101 Mohandas, E.; Rajmohan, V.; Raghunath, B.; Indian J. Psychiatry2009 , 51, 55. Por outro lado, o sistema colinérgico possui a capacidade de exercer função reguladora sobre o processamento do peptídeo amiloide.100100 Parikh, V.; Bernard, C. S.; Naughton, S. X.; Yegla, B.;Behav. Brain Res.2014 , 274, 30.,102102 Kar, S.; Slowikowski, S. P.; Westaway, D.; Mount, H. T.; J. Psychiatry Neurosci.2004 , 29, 427. Portanto, é natural supor que ambos os aspectos possam apresentar influência mútua, tornando difícil entender qual seria o evento desencadeador da patologia.100100 Parikh, V.; Bernard, C. S.; Naughton, S. X.; Yegla, B.;Behav. Brain Res.2014 , 274, 30.,103103 Yan, Z.; Feng, J.; Curr. Alzheimer Res.2004 , 1, 241. Tem sido observado que a atividade colinérgica é capaz de influenciar o processamento amiloide: na ausência da atividade do receptor muscarínico é privilegiado o caminho amiloidogênico, enquanto acontece o contrário com a atividade receptora normal.103103 Yan, Z.; Feng, J.; Curr. Alzheimer Res.2004 , 1, 241.,104104 Auld, D. S.; Kornecook, T. J.; Bastianetto, S.; Quirion, R.;Prog. Neurobiol.2002 , 68, 209. Para continuar estudando esta hipótese, são necessários modelos animais confiáveis, que repliquem a depleção colinérgica no córtex e no hipocampo e a presença difusa, no tecido cerebral, das placas amiloides e das massas neurofibrilares presentes em pacientes com DA.
A hipótese metálica
Nos últimos anos, com a ampla discussão a respeito da hipótese da cascata amiloide, um número crescente de evidências passou a sugerir que os íons metálicos endógenos, particularmente os que possuem atividade redox, tais como cobre(II) e ferro(III), além de certos íons não redox-ativos, como o zinco(II), podem contribuir na evolução de doenças neurodegenerativas, favorecendo a agregação de Aβ e aumentando a sua toxicidade.6060 Hane, F.; Leonenko, Z.; Biomolecules2014 , 4, 101.,6565 Hane, F.; Tran, G.; Attwood, S. J.; Leonenko, Z.; PLoS One2013 , 8, e59005.,105105 Deibel, M. A.; Ehmann, W. D.; Markesbery, W. R.; J. Neurol. Sci.1996 , 143, 137.
106 Azimi, S.; Rauk, A.; Int. J. Alzheimer's Dis.2011 , 2011, 539762.
107 Tõugu, V.; Karafin, A.; Palumaa, P.; J. Neurochem.2008 , 104, 1249.
108 Huang, X.; Atwood, C. S.; Moir, R. D.; Hartshorn, M. A.; Tanzi, R. E.; Bush, A. I.; J. Biol. Inorg. Chem.2004 , 9, 954.-109109 Craddock, T. J.; Tuszynski, J. A.; Chopra, D.; Casey, N.; Goldstein, L. E.; Hameroff, S. R.; Tanzi, R. E.; PLoS One2012 , 7, e33552.
Por exemplo, tanto o cobre quanto o zinco aumentam a velocidade de agregação do peptídeo Aβ sintético em meio aquoso,110110 Miura, T.; Suzuki, K.; Kohata, N.; Takeuchi, H.;Biochemistry2000 , 39, 7024.,111111 Bush, A. I.; Pettingell, W. H.; Multhaup, G.; d Paradis, M.; Vonsattel, J. P.; Gusella, J. F.; Beyreuther, K.; Masters, C. L.; Tanzi, R. E.;Science1994 , 265, 1464.provavelmente por ligação aos resíduos de histidina do mesmo,112112 Huang, X.; Atwood, C. S.; Moir, R. D.; Hartshorn, M. A.; Vonsattel, J. P.; Tanzi, R. E.; Bush, A. I.; J. Biol. Chem.1997 , 272, 26464.,113113 Yang, D. S.; McLaurin, J.; Qin, K.; Westaway, D.; Fraser, P. E.;Eur. J. Biochem.2000 , 267, 6692. e estão anormalmente distribuídos em pacientes com DA,114114 Smith, M. A.; Wehr, K.; Harris, P. L.; Siedlak, S. L.; Connor, J. R.; Perry, G.; Brain Res.1998 , 788, 232.,115115 Faller, P.; Hureau, C.; Chemistry2012 , 18, 15910. particularmente presentes no hipocampo e nas amídalas, no interior do núcleo e nas áreas periféricas das placas senis.105105 Deibel, M. A.; Ehmann, W. D.; Markesbery, W. R.; J. Neurol. Sci.1996 , 143, 137.,116116 Danscher, G.; Jensen, K. B.; Frederickson, C. J.; Kemp, K.; Andreasen, A.; Juhl, S.; Stoltenberg, M.; Ravid, R.; J. Neurosci. Methods1997 , 76, 53.
117 Lovell, M. A.; Robertson, J. D.; Teesdale, W. J.; Campbell, J. L.; Markesbery, W. R.; J. Neurol. Sci.1998 , 158, 47.-118118 Sayre, L. M.; Perry, G.; Harris, P. L.; Liu, Y.; Schubert, K. A.; Smith, M. A.; J. Neurochem.2000 , 74, 270.
Estes biometais induzem o aumento do estresse oxidativo no cérebro, devido à sua capacidade de produzir espécies reativas de oxigênio (EROs), como radicais hidroxila e peróxido de hidrogênio, além de espécies reativas de nitrogênio (ERNs), como o óxido nítrico, via reações de Haber-Weiss e de Fenton (Esquema 1). Os danos celulares causados pelas EROs e ERNs são extensos. Por exemplo, a oxidação do ferro por meio da reação de Fenton gera anormalidades no RNA, que, na DA, é particularmente afetado, causando grande redução na síntese protéica,119119 Zhu, X.; Su, B.; Wang, X.; Smith, M. A.; Perry, G.; Cell. Mol. Life Sci.2007 , 64, 2202.,120120 Bonda, D. J.; Lee, H. G.; Blair, J. A.; Zhu, X.; Perry, G.; Smith, M. A.; Metallomics2011 , 3, 267.enquanto o radical hidroxila provoca diversos danos às biomoléculas atacando as bases nitrogenadas e a desoxirribose do DNA, reagindo com as cadeias laterais de aminoácidos e proteínas (podendo gerar fragmentos protéicos não funcionais) e também com lipídeos de membrana, convertendo sítios lipídicos específicos em novos centros de formação de radicais livres.121121 Barreiros, A. L. B. S.; David, J. M.; David, J. P.; Quim. Nova2006 , 29, 113.
122 Barnham, K. J.; Bush, A. I.; Curr. Opin. Chem. Biol.2008 , 12, 222.-123123 Barnham, K. J.; Masters, C. L.; Bush, A. I.; Nat. Rev. Drug Discov.2004 , 3, 205.
Uma abordagem mais ampla especula sobre o fato da secreção e a deposição de Aβ, assim como a agregação das placas, ocorrerem como consequência do estresse oxidativo.124124 Su, B.; Wang, X.; Nunomura, A.; Moreira, P. I.; Lee, H.; Perry, G.; Smith, M. A.; Zhu, X.; Curr. Alzheimer Res.2008 , 5, 525.,125125 Liu, G.; Men, P.; Kudo, W.; Perry, G.; Smith, M. A.;Neurosci. Lett.2009 , 455, 187. Este enfoque assume a hipótese das placas amiloides não serem unicamente a causa da doença, mas, também, contribuírem para a desregulação da homeostase metálica nas sinapses.126126 Adlard, P. A.; Parncutt, J. M.; Finkelstein, D. I.; Bush, A. I.;J. Neurosci.2010 , 30, 1631.,127127 Lee, J. Y.; Cole, T. B.; Palmiter, R. D.; Suh, S. W.; Koh, J. Y.;Proc. Natl. Acad. Sci. U. S. A.2002 , 99, 7705. Um importante indício a favor dessa abordagem foi a demonstração de que o zinco liberado na sinapse glutamatérgica, necessário para manter a memória e a cognição, tem o seu transportador ZnT3 expresso em maiores quantidades em áreas afetadas pela patologia amiloide.126126 Adlard, P. A.; Parncutt, J. M.; Finkelstein, D. I.; Bush, A. I.;J. Neurosci.2010 , 30, 1631. Reforçando estes resultados, foi observado também que o transporte de zinco nas vesículas sinápticas é coincidente com a expressão de ZnT3 e encontra-se completamente ausente em ratos transgênicos apresentando deficiência desse transportador.128128 Cole, T. B.; Wenzel, H. J.; Kafer, K. E.; Schwartzkroin, P. A.; Palmiter, R. D.; Proc. Natl. Acad. Sci. U. S. A.1999 , 96, 1716. Além disso, foi demonstrado que o zinco liberado na sinapse glutamatérgica contribui predominantemente na deposição das placas amiloides em ratos transgênicos expressando a patologia amiloide, e que a perda de ZnT3 inibe a liberação dos íons Zn2+, levando ao declínio cognitivo, corroborando ainda mais esta hipótese.126126 Adlard, P. A.; Parncutt, J. M.; Finkelstein, D. I.; Bush, A. I.;J. Neurosci.2010 , 30, 1631.,127127 Lee, J. Y.; Cole, T. B.; Palmiter, R. D.; Suh, S. W.; Koh, J. Y.;Proc. Natl. Acad. Sci. U. S. A.2002 , 99, 7705. Como os níveis de ZnT3 diminuem com a idade, e este decréscimo é ainda mais significativo em casos de DA, tem sido proposto que a diminuição idade-dependente do movimento trans-sináptico do zinco seja responsável pela perda cognitiva, justificando esse evento pelo fato das placas de Aβ serem agregadas covalentemente por este metal. Assim, a lesão amiloide da DA poderia causar perdas cognitivas ao sequestrar o zinco extracelular.109109 Craddock, T. J.; Tuszynski, J. A.; Chopra, D.; Casey, N.; Goldstein, L. E.; Hameroff, S. R.; Tanzi, R. E.; PLoS One2012 , 7, e33552.,126126 Adlard, P. A.; Parncutt, J. M.; Finkelstein, D. I.; Bush, A. I.;J. Neurosci.2010 , 30, 1631.,129129 Bush, A. I.; J. Alzheimer's Dis.2013 , 33, S277.
130 Miller, L. M.; Wang, Q.; Telivala, T. P.; Smith, R. J.; Lanzirotti, A.; Miklossy, J.; J. Struct. Biol.2006 , 155, 30.-131131 Miller, Y.; Ma, B.; Nussinov, R.; Proc. Natl. Acad. Sci. U. S. A.2010 , 107, 9490.
Diversos estudos revelam que zinco e cobre competem pelos mesmos resíduos de Aβ, tendo o zinco uma maior relevância na rápida agregação do peptídeo do que o cobre, o qual, por sua vez, induz principalmente mudanças conformacionais no peptídeo.132132 Hoernke, M.; Koksch, B.; Brezesinski, G.; Biophys. Chem.2010 , 150, 64.,133133 Marino, T.; Russo, N.; Toscano, M.; Pavelka, M.;Interdiscip. Sci.2010 , 2, 57. Esses dados mostram a importância da determinação de metais em amostras biológicas relacionadas à DA e também do uso de terapias de quelação.134134 Leal, M. F. C.; Catarino, R. I. L.; Pimenta, A. M.; Souto, M. R. S.; Pinheiro, T. S. N.; Quim. Nova2012 , 35, 1985. Por exemplo, os trabalhos do grupo de pesquisa liderado por A. Bush sugerem que a desregulação exacerbada da homeostase metálica não levaria somente à agregação e deposição de Aβ, mas que, em paralelo, conduziria ao acúmulo de ferro dentro dos neurônios, causando danos oxidativos e neurodegeneração.111111 Bush, A. I.; Pettingell, W. H.; Multhaup, G.; d Paradis, M.; Vonsattel, J. P.; Gusella, J. F.; Beyreuther, K.; Masters, C. L.; Tanzi, R. E.;Science1994 , 265, 1464.,123123 Barnham, K. J.; Masters, C. L.; Bush, A. I.; Nat. Rev. Drug Discov.2004 , 3, 205.,126126 Adlard, P. A.; Parncutt, J. M.; Finkelstein, D. I.; Bush, A. I.;J. Neurosci.2010 , 30, 1631.,129129 Bush, A. I.; J. Alzheimer's Dis.2013 , 33, S277.,135135 Adlard, P. A.; Bica, L.; White, A. R.; Nurjono, M.; Filiz, G.; Crouch, P. J.; Donnelly, P. S.; Cappai, R.; Finkelstein, D. I.; Bush, A. I.;PLoS One2011 , 6, e17669.
136 Friedlich, A. L.; Lee, J. Y.; van Groen, T.; Cherny, R. A.; Volitakis, I.; Cole, T. B.; Palmiter, R. D.; Koh, J. Y.; Bush, A. I.; J. Neurosci.2004 , 24, 3453.-137137 Sensi, S. L.; Paoletti, P.; Koh, J. Y.; Aizenman, E.; Bush, A. I.; Hershfinkel, M.; J. Neurosci.2011 , 31, 16076. Este grupo também explorou a interação funcional dos sistemas de transporte de metal e proteínas envolvidas na neurodegeneração, avaliando a hipótese destas proteínas serem desnaturadas na presença de metais, devido ao fato das mesmas falharem no papel de regulação destes, por mecanismo de retroação negativa, resultando na sobrecarga das próprias pelo acúmulo dos metais. Estes estudos apresentam um mecanismo de toxicidade inédito, intrínseco ao processo de formação de Aβ, definindo uma relação patológica entre o acúmulo de zinco extracelular nas placas amiloides e o acúmulo de ferro intraneuronal observado na DA. Na teoria de Bush e colaboradores,129129 Bush, A. I.; J. Alzheimer's Dis.2013 , 33, S277. a partir do momento em que o ferro se liga aos emaranhados neurofibrilares, induzindo o estresse oxidativo,138138 Smith, M. A.; Harris, P. L.; Sayre, L. M.; Perry, G.; Proc. Natl. Acad. Sci. U. S. A.1997 , 94, 9866. a proteína tau poderia apresentar papel fisiológico na homeostase neuronal do ferro, pois ela direciona o tráfego de β-APP para a superfície dos neurônios,139139 Lei, P.; Ayton, S.; Finkelstein, D. I.; Spoerri, L.; Ciccotosto, G. D.; Wright, D. K.; Wong, B. X.; Adlard, P. A.; Cherny, R. A.; Lam, L. Q.; Roberts, B. R.; Volitakis, I.; Egan, G. F.; McLean, C. A.; Cappai, R.; Duce, J. A.; Bush, A. I.; Nat. Med.2012 , 18, 291. para interagir com a ferroportina.
Neste contexto, o envolvimento de biometais na patogênese da DA vem cada vez mais sendo estudado, tanto devido à sua capacidade de causar estresse oxidativo, quanto pela sua participação na secreção, no transporte e no depósito do peptídeo Aβ, dentre outros.
Não apenas os metais fisiológicos têm sido implicados na patogênese desta doença, mas também alguns metais considerados tóxicos, tais como o alumínio, o chumbo e o mercúrio. Por outro lado, apesar de o selênio ser essencial a baixíssimas concentrações, optamos por discutir a sua relação com a DA conjuntamente com os metais tóxicos.
Diversos estudos indicam que existe uma relação direta entre o alumínio e a DA. Este metal tem sido observado em maiores concentrações no cérebro de pacientes acometidos pela doença, e existem relatos de riscos estatisticamente aumentados em populações expostas a níveis de alumínio acima de 0,1 mg L-1 na água potável.140140 Shcherbatykh, I.; Carpenter, D. O.; J. Alzheimer's Dis.2007 , 11, 91.,141141 Frisardi, V.; Solfrizzi, V.; Capurso, C.; Kehoe, P. G.; Imbimbo, B. P.; Santamato, A.; Dellegrazie, F.; Seripa, D.; Pilotto, A.; Capurso, A.; Panza, F.; J. Alzheimer's Dis.2010 , 20, 17. O alumínio é absorvido em maiores quantidades por pessoas e animais mais velhos, o que leva a uma maior susceptibilidade dos indivíduos idosos ao acúmulo deste elemento no cérebro.142142 Bakulski, K. M.; Rozek, L. S.; Dolinoy, D. C.; Paulson, H. L.; Hu, H.; Curr. Alzheimer Res.2012 , 9, 563. Em pacientes com DA, foi demonstrada uma absorção três vezes maior de alumínio em comparação com pessoas sem a doença.143143 von Lindern, I.; Spalinger, S.; Petroysan, V.; von Braun, M.;Sci. Total Environ.2003 , 303, 139. Estudos em cobaias indicam que o alumínio modifica estruturas cerebrais, como, por exemplo, o número ou a distribuição de cargas presentes nas superfícies do cérebro, com consequente alteração da atividade da barreira hematoencefálica,144144 Lewin, M. D.; Sarasua, S.; Jones, P. A.; Environ. Res.1999 , 81, 52. e que a exposição crônica a este elemento por meio da ingestão de água potável aumenta processos inflamatórios no cérebro.145145 Brown, D. R.; Dalton Trans.2009 , 4069. Diversos mecanismos de ação têm sido propostos para explicar a toxicidade do alumínio na DA. Um deles envolve a sua interação com a calmodulina, uma proteína moduladora de várias proteínas e enzimas que se liga ao Ca2+, inibindo a inativação Ca-dependente do canal receptor de NMDA,146146 Levi, R.; Wolf, T.; Fleminger, G.; Solomon, B.; Mol. Cell. Biochem.1998 , 189, 41. enquanto seu envolvimento na ativação indireta de serino-proteases, como a α-quimiotripsina, também tem sido citado, com o consequente aumento no processamento da APP, acúmulo do peptídeo Aβ e formação das placas amiloides.147147 Kawahara, M.; Kato-Negishi, M.; Int. J. Alzheimer's Dis.2011 , 2011, 1. Além disso, foi demonstrado que o alumínio também altera a atividade da AChE,148148 Zatta, P.; Ibn-Lkhayat-Idrissi, M.; Zambenedetti, P.; Kilyen, M.; Kiss, T.; Brain Res. Bull.2002 , 59, 41. liga-se às porções polares das membranas celulares, prejudicando processos de transporte e o metabolismo celular e promove estresse oxidativo no cérebro, ao acelerar a peroxidação de membranas lipídicas na presença de Fe2+ e também modificar a homeostase deste metal.142142 Bakulski, K. M.; Rozek, L. S.; Dolinoy, D. C.; Paulson, H. L.; Hu, H.; Curr. Alzheimer Res.2012 , 9, 563.,147147 Kawahara, M.; Kato-Negishi, M.; Int. J. Alzheimer's Dis.2011 , 2011, 1. O alumínio também apresenta efeitos tóxicos no retículo endoplasmático e nas mitocôndrias, induzindo a apoptose das células neuronais,149149 Savory, J.; Herman, M. M.; Ghribi, O.; J. Alzheimer's Dis.2006 , 10, 135. o que certamente contribui para a progressiva perda neuronal observada na DA. Interessantemente, esta cascata de eventos apoptóticos não foi verificada para outros metais.147147 Kawahara, M.; Kato-Negishi, M.; Int. J. Alzheimer's Dis.2011 , 2011, 1.
Estudos recentes indicam que a exposição de roedores e primatas a chumbo na infância causa modificações na expressão de genes e biomarcadores relacionados à DA quando os animais atingem a idade adulta. Foram observadas a superexpressão de APP e BACE1, a presença de placas senis com alterações da distribuição intracelular do peptídeo Aβ, aumento dos níveis de metilação e danos oxidativos no DNA.150150 Basha, M. R.; Wei, W.; Bakheet, S. A.; Benitez, N.; Siddiqi, H. K.; Ge, Y. W.; Lahiri, D. K.; Zawia, N. H.; J. Neurosci.2005 , 25, 823.
151 Wu, J.; Basha, M. R.; Brock, B.; Cox, D. P.; Cardozo-Pelaez, F.; McPherson, C. A.; Harry, J.; Rice, D. C.; Maloney, B.; Chen, D.; Lahiri, D. K.; Zawia, N. H.; J. Neurosci.2008 , 28, 3.-152152 Bolin, C. M.; Basha, R.; Cox, D.; Zawia, N. H.; Maloney, B.; Lahiri, D. K.; Cardozo-Pelaez, F.; FASEB J.2006 , 20, 788.
O mercúrio é um metal extremamente tóxico em todas as suas formas, e também tem sido alvo de hipóteses relacionadas com a DA. Por exemplo, a exposição a mercúrio em ratos causou a inibição da guanosina trifosfato à tubulina nos cérebros das cobaias, inibindo por sua vez a polimerização da tubulina em microtúbulos.153153 Pendergrass, A. C.; Haley, B. E.; Vimy, M. J.; Winfield, S. A.; Lorscheider, F. L.; Neurotoxicology1997 , 18, 315. Isto identifica uma relação importante entre este metal e a DA, pois este tipo de lesão molecular é extremamente semelhante ao observado em 80% dos cérebros de pacientes com DA. Outros estudos demonstraram que a presença de mercúrio também modifica os padrões de crescimento de células neuríticas in vitro, desintegrando a estrutura tubulina/microtúbulo, formando, também, agregados neurofibrilares.154154 Leong, C. C. W.; Syed, N. I.; Lorscheider, F. L.;Neuroreport2001 , 12, 733. Alguns autores também postulam que, na presença de outros metais como zinco, cádmio e chumbo, o mercúrio apresenta toxicidade exacerbada devido a mecanismos sinergísticos, levando a crer que não é necessário que o mercúrio esteja presente em níveis elevados no cérebro de pacientes com DA para ser considerado causal na etiologia da doença.155155 Haley, B. E.; Medical Veritas2007 , 4, 1510. Além disso, existe também a hipótese de que a susceptibilidade à DA de pessoas contendo o apolipoproteína (ApoE) gene possa estar relacionada com a contaminação por este metal.155155 Haley, B. E.; Medical Veritas2007 , 4, 1510.
O selênio é um micronutriente importante para a manutenção da saúde humana, que age como antioxidante combinado à glutationa peroxidase, uma enzima que atua como um mecanismo de defesa contra radicais livres endógenos. O selênio também se liga a aminoácidos para formar pequenos peptídeos chamados selenoproteínas, que exercem atividades antioxidantes.156156 Santos, J. R.; Gois, A. M.; Mendonça, D. M.; Freire, M. A.;Front Aging Neurosci.2014 , 6, 206.A relevância do selênio na DA se deve ao seu importante papel antioxidante, visto que a doença é caracterizada por vasto estresse oxidativo no cérebro. Recentemente, em 2015, Cardoso e colaboradores publicaram uma revisão a respeito do papel do selênio no contexto da DA, citando diversos estudos conduzidos para tentar compreender o papel deste elemento na patologia da doença. A homeostase do selênio parece ser parcialmente desregulada na DA.157157 Cardoso, B. R.; Roberts, B. R.; Bush, A. I.; Hare, D. J.;Metallomics2015 , 7, 1213. Estudos em humanos reportaram correlação negativa entre o declínio cognitivo e os níveis de selênio.158158 González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J. L.;Biometals2014 , 27, 539. Há uma diminuição nos níveis de selênio de eritrócitos em idosos com MCI (do inglês, Mild Cognitive Impairment ) e DA em comparação com controles, que está correlacionado com um decréscimo da função cognitiva. Entretanto, não foi reportada diferença significativa entre os níveis de selênio no plasma de indivíduos sadios e apresentando MCI.159159 Rita Cardoso, B.; Silva Bandeira, V.; Jacob-Filho, W.; Franciscato Cozzolino, S. M.; J. Trace Elem. Med. Biol.2014 , 28, 422. Uma vez que o plasma indica exposição recente e os eritrócitos se referem ao status do selênio a longo prazo, estes resultados sugerem que deficiência crônica está correlacionada com declínio cognitivo.157157 Cardoso, B. R.; Roberts, B. R.; Bush, A. I.; Hare, D. J.;Metallomics2015 , 7, 1213. Em pacientes acometidos pela DA que apresentam MCI, os níveis de selênio no plasma são mais baixos quando comparados com idosos saudáveis de mesma idade.160160 Olde Rikkert, M. G.; Verhey, F. R.; Sijben, J. W.; Bouwman, F. H.; Dautzenberg, P. L.; Lansink, M.; Sipers, W. M.; van Asselt, D. Z.; van Hees, A. M.; Stevens, M.; Vellas, B.; Scheltens, P.; J. Alzheimer's Dis.2014 , 41, 261. Estes estudos indicam que a falta de selênio pode aumentar o risco de demência. Estudos com modelos animais transgênicos mostraram que uma dieta deficiente em selênio está associada com o aumento da formação de placas senis em cérebros de ratos.161161 Haratake, M.; Yoshida, S.; Mandai, M.; Fuchigami, T.; Nakayama, M.;Metallomics2013 , 5, 479. O tratamento com selenato de sódio reduz a fosforilação da proteína tau tanto in vitro quanto em modelos de animais transgênicos.162162 van Eersel, J.; Ke, Y. D.; Liu, X.; Delerue, F.; Kril, J. J.; Götz, J.; Ittner, L. M.; Proc. Natl. Acad. Sci. U. S. A.2010 , 107, 13888.,163163 Corcoran, N. M.; Martin, D.; Hutter-Paier, B.; Windisch, M.; Nguyen, T.; Nheu, L.; Sundstrom, L. E.; Costello, A. J.; Hovens, C. M.; J. Clin. Neurosci.2010 , 17, 1025. Derivados da 8-hidroxiquinolina contendo selênio ligados a Cu2+, Fe2+ e Zn2+ têm demonstrado capacidade de inibição da agregação e também a dissociação de agregados de Aβ induzidos por Cu2+.164164 Wang, Z.; Wang, Y.; Li, W.; Mao, F.; Sun, Y.; Huang, L.; Li, X.;ACS Chem. Neurosci.2014 , 5, 952.
A hipótese do diabetes de tipo 3
O cérebro humano é, do ponto de vista metabólico, um dos órgãos mais ativos do nosso corpo, processando uma grande quantidade de carboidratos para produzir energia celular na forma de adenosina trifosfato (ATP). Apesar das suas exigências, o cérebro não possui uma grande flexibilidade em termos de substratos para a produção desta energia, baseando-se quase exclusivamente na utilização de glicose. Esta dependência põe em risco o órgão, caso o fornecimento do substrato seja escasso ou interrompido, ou caso a capacidade de metabolizar a glicose se torne falha: o cérebro se torna incapaz de proteger as sinapses. Nesta situação, as células podem não funcionar corretamente, resultando em alterações cognitivas. A partir deste princípio básico, torna-se evidente uma possível ligação entre o diabetes e a DA.165165 Ferreira, S. T.; Clarke, J. R.; Bomfim, T. R.; De Felice, F. G.;Alzheimer's Dementia2014 , 10, S76.,166166 Lourenco, M. V.; Ferreira, S. T.; De Felice, F. G.; Prog. Neurobiol.2015 , 129, 37.
A pesquisa sobre a relação entre o diabetes e a DA começou com o chamado "estudo Rotterdam", um estudo epidemiológico que investigou mais de 6000 idosos por dois anos e apontou uma correlação positiva entre a presença de diabetesmellitus e o desenvolvimento de demência.167167 Ott, A.; Stolk, R. P.; van Harskamp, F.; Pols, H. A.; Hofman, A.; Breteler, M. M.; Neurology1999 , 53, 1937. Outro trabalho epidemiológico, mais recente, mostrou a incidência de aumento da DA em homens que ganharam peso entre os 30 e 45 anos de idade e em mulheres entre 30 e 45 anos com índice de massa corpórea maior que 30.168168 Beydoun, M. A.; Lhotsky, A.; Wang, Y.; Dal Forno, G.; An, Y.; Metter, E. J.; Ferrucci, L.; O'Brien, R.; Zonderman, A. B.; Am. J. Epidemiol.2008 , 168, 1179. Já um estudo sueco apontou o aumento estatisticamente significativo do risco de homens que desenvolvem diabetes tipo 2 por volta dos 50 anos de idade de desenvolverem a DA. Pesquisadores também verificaram que homens com baixa produção de insulina aos 50 anos apresentaram 150% a mais de probabilidade de desenvolver DA do que aqueles com produção normal de insulina. Esta associação foi ainda maior em pacientes com deficiência da apolipoproteína E4 (ApoE4), o que parece indicar uma forte predisposição genética para a DA, tornando assim o diabetes um possível fator de risco independente para esta doença.169169 Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L.;Neurology2008 , 71, 1065. As apolipoproteínas (Apo) são proteínas plasmáticas, com a capacidade de se ligar a lipídeos e com a função de transportar triglicerídeos e colesterol para os órgãos, sendo o colesterol liberado utilizado para apoiar a manutenção sinaptogênica e das conexões sinápticas.170170 Pfrieger, F. W.; Cell. Mol. Life Sci.2003 , 60, 1158. A ApoE humana é uma glicoproteína composta por 299 aminoácidos e que apresenta níveis variáveis de modificação pós-traducional.171171 Wernette-Hammond, M. E.; Lauer, S. J.; Corsini, A.; Walker, D.; Taylor, J. M.; Rall, S. C.; J. Biol. Chem.1989 , 264, 9094. Alguns estudos histopatológicos observaram uma correlação positiva entre a densidade das placas senis e a quantidade de ApoE ε4 no cérebro de pacientes com DA,172172 Rebeck, G. W.; Reiter, J. S.; Strickland, D. K.; Hyman, B. T.;Neuron1993 , 11, 575.,173173 Schmechel, D. E.; Saunders, A. M.; Strittmatter, W. J.; Crain, B. J.; Hulette, C. M.; Joo, S. H.; Pericak-Vance, M. A.; Goldgaber, D.; Roses, A. D.; Proc. Natl. Acad. Sci. U. S. A.1993 , 90, 9649. e uma pesquisa recente abrangendo uma população de estudo maior sugeriu fortemente que a deficiência de ApoEε4 encontra-se associada ao aumento das placas neuríticas.174174 Tiraboschi, P.; Hansen, L. A.; Masliah, E.; Alford, M.; Thal, L. J.; Corey-Bloom, J.; Neurology2004 , 62, 1977. Outros estudos, porém, não observaram nenhum tipo de relação entre esses parâmetros.175175 Benjamin, R.; Leake, A.; Ince, P. G.; Perry, R. H.; McKeith, I. G.; Edwardson, J. A.; Morris, C. M.; Neurodegen.1995 , 4, 443.
176 Heinonen, O.; Lehtovirta, M.; Soininen, H.; Helisalmi, S.; Mannermaa, A.; Sorvari, H.; Kosunen, O.; Paljärvi, L.; Ryynänen, M.; Riekkinen, P. J.; J. Neurobiol. Aging1995 , 16, 505.
177 Itoh, Y.; Yamada, M.; N. Engl. J. Med.1996 , 334, 599.-178178 Landén, M.; Thorsell, A.; Wallin, A.; Blennow, K.; J. Neurol., Neurosurg. Psychiatry1996 , 61, 352. Existem diferentes hipóteses sobre a influência desta proteína na patogênese da DA, sendo a principal baseada na função de ApoE como uma proteína que se liga ao Aβ, induzindo mudanças conformacionais patológicas no peptídeo, acelerando a sua deposição no cérebro.179179 Wisniewski, T.; Frangione, B.; Neurosci. Lett.1992 , 135, 235. Com base nesta hipótese, estudos envolvendo uma população de adultos de meia idade com o genótipo ApoE ε4, porém não apresentando problemas de cognição, foram recentemente realizados e os resultados sugeriram que a deposição do peptídeo Aβ parece iniciar-se mais cedo nesses indivíduos.180180 Reiman, E. M.; Chen, K.; Liu, X.; Bandy, D.; Yu, M.; Lee, W.; Ayutyanont, N.; Keppler, J.; Reeder, S. A.; Langbaum, J. B.; Alexander, G. E.; Klunk, W. E.; Mathis, C. A.; Price, J. C.; Aizenstein, H. J.; DeKosky, S. T.; Caselli, R. J.; Proc. Natl. Acad. Sci. U. S. A.2009 , 106, 6820.,181181 Sunderland, T.; Mirza, N.; Putnam, K. T.; Linker, G.; Bhupali, D.; Durham, R.; Soares, H.; Kimmel, L.; Friedman, D.; Bergeson, J.; Csako, G.; Levy, J. A.; Bartko, J. J.; Cohen, R. M.; Biol. Psychiatry2004 , 56, 670.
A terminologia "diabetes de tipo 3" foi introduzida em 2005 por Suzanne de la Monte, cujo grupo de pesquisa examinou o tecido cerebral de pacientes com DA que vieram a óbito, observando que a patologia demonstra elementos dos diabetes de tipo 1 e 2, ou seja, além da diminuição na produção de insulina, é também observada a resistência dos receptores da insulina, sugerindo que a DA pode ser uma doença neuroendócrina associada à sinalização deste hormônio.182182 Steen, E.; Terry, B. M.; Rivera, E. J.; Cannon, J. L.; Neely, T. R.; Tavares, R.; Xu, X. J.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2005 , 7, 63.,183183 Rivera, E. J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2005 , 8, 247.
Os estudos acima descritos verificaram que a expressão da insulina se mostrou inversamente proporcional ao estágio Braak da doença,184184 Braak, H.; Braak, E.; Acta Neuropathol.1991 , 82, 239. com uma redução de 80% no número de receptores de insulina em pacientes com DA, em comparação com indivíduos normais. No cenário do diagnóstico da DA, os estágios da doença são determinados segundo a escala de Braak, que avalia, principalmente, os sintomas clínicos de demência em correlação com a distribuição de emaranhados neurofibrilares no cérebro. Os estágios são definidos como I e II quando o envolvimento dos emaranhados está confinado principalmente à região trans-entorrinal do cérebro, traduzido sintomatologicamente como níveis de cognição normais e levemente afetadas, respectivamente. Já nas fases III e IV, há também o envolvimento das regiões límbicas, como o hipocampo, levando ao prejuízo cognitivo de leve a moderado, de confusão e perda de memória, desorientação, problemas com tarefas cotidianas, mudanças na personalidade e na capacidade de julgamento no estágio III e sintomas psicóticos como ansiedade, desconfiança e agitação, além de distúrbios do sono no estágio IV. Por último, nos estágios V e VI, existe grande envolvimento neocortical, acarretando a dificuldade em reconhecer familiares e amigos, perda da fala, do apetite e do controle da bexiga e do intestino. Neste contexto, a capacidade da insulina de se ligar aos seus receptores se mostrou comprometida, com redução nos níveis de RNA mensageiro correspondente à insulina e seus receptores, observando-se também redução nos níveis da proteína tau.183183 Rivera, E. J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2005 , 8, 247.,185185 de la Monte, S. M.; Tong, M.; Lester-Coll, N.; Plater, M.; Wands, J. R.; J. Alzheimer's Dis.2006 , 10, 89.,186186 De Felice, F. G.; Lourenco, M. V.; Ferreira, S. T.;Alzheimer's Dementia2014 , 10, S26.Estes resultados têm inspirado estudos em modelos animais, em que a injeção intracerebral de estreptozotocina, uma droga para a indução de diabetes em ratos, além de resultar na degradação da insulina e da alteração dos mecanismos de sinalização do fator de crescimento semelhante à insulina, desencadeou o aparecimento de lesões oxidativas no cérebro das cobaias. A combinação destas alterações derivou finalmente em neurodegeneração, incluindo certas características neurológicas típicas da DA.187187 Lester-Coll, N.; Rivera, E. J.; Soscia, S. J.; Doiron, K.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2006 , 9, 13.
A insulina, importante no processamento da memória, tem a capacidade de atravessar a barreira hematoencefálica e é produzida constitutivamente no tecido cerebral. De forma geral, pacientes com DA apresentam diminuição da concentração de insulina e menor número de receptores da mesma. Quando estes sintomas são corrigidos farmacologicamente, uma melhora nos processos cognitivos dos pacientes é observada. A insulina liga-se a receptores específicos no cérebro, a maioria dos quais estão localizados no córtex cerebral, hipocampo, bulbo olfatório, cerebelo e hipotálamo. Devido ao fato destes receptores se localizarem nas áreas do cérebro pertinentes à cognição, é válido considerar a associação entre a insulina e a cognição.188188 Craft, S.; Watson, G. S.; Lancet Neurol.2004 , 3, 169. Diversos estudos utilizando a administração de insulina intranasal, intravenosa e intracerebral demonstraram uma melhoria na cognição das cobaias analisadas.188188 Craft, S.; Watson, G. S.; Lancet Neurol.2004 , 3, 169.
189 Reger, M. A.; Watson, G. S.; Green, P. S.; Wilkinson, C. W.; Baker, L. D.; Cholerton, B.; Fishel, M. A.; Plymate, S. R.; Breitner, J. C.; DeGroodt, W.; Mehta, P.; Craft, S.; Neurology2008 , 70, 440.-190190 Park, C. R.; Seeley, R. J.; Craft, S.; Woods, S. C.;Physiol. Behav.2000 , 68, 509.
Evidências experimentais suportam a ideia de que os efeitos tóxicos de Aβ podem promover resistência à insulina. Assim, estes resultados sugerem o desenvolvimento de uma retroalimentação positiva na neurodegeneração progressiva, isto é, a resistência à insulina levaria ao acúmulo de Aβ e a toxicidade deste peptídeo determinaria a resistência à insulina no cérebro.191191 Bomfim, T. R.; Forny-Germano, L.; Sathler, L. B.; Brito-Moreira, J.; Houzel, J. C.; Decker, H.; Silverman, M. A.; Kazi, H.; Melo, H. M.; McClean, P. L.; Holscher, C.; Arnold, S. E.; Talbot, K.; Klein, W. L.; Munoz, D. P.; Ferreira, S. T.; De Felice, F. G.; J. Clin. Invest.2012 , 122, 1339.,192192 Talbot, K.; Wang, H. Y.; Kazi, H.; Han, L. Y.; Bakshi, K. P.; Stucky, A.; Fuino, R. L.; Kawaguchi, K. R.; Samoyedny, A. J.; Wilson, R. S.; Arvanitakis, Z.; Schneider, J. A.; Wolf, B. A.; Bennett, D. A.; Trojanowski, J. Q.; Arnold, S. E.; J. Clin. Invest.2012 , 122, 1316. Esse fenômeno poderia explicar porque a mensuração dos níveis de Aβ no líquor ou a análise por imagem do cérebro têm se mostrado inadequadas como biomarcadores únicos para o diagnóstico definitivo da DA, e porque os resultados de ensaios clínicos de terapia anti-Aβ têm se mostrado inconclusivos até o momento.9393 Ferreira, S. T.; Klein, W. L.; Neurobiol. Learn. Mem.2011 , 96, 529.,186186 De Felice, F. G.; Lourenco, M. V.; Ferreira, S. T.;Alzheimer's Dementia2014 , 10, S26.,193193 de la Monte, S. M.; Drugs2012 , 72, 49.
A evolução das hipóteses etiológicas para a DA
Para observar a evolução da produção científica a respeito das diferentes teorias envolvendo as bases moleculares da DA ao longo do tempo, conduziu-se uma pesquisa bibliográfica a respeito. Os resultados foram obtidos a partir de buscas na base de dados PubMedTM em Junho de 2015. A base de dados PubMed é uma plataforma de pesquisa que cataloga, primariamente, informações a respeito de tópicos médicos e biomédicos. A pesquisa foi realizada utilizando-se palavras-chave específicas para cada teoria, conforme descrito abaixo, no período de 1980 a 2014. Os resultados foram subsequentemente refinados para fornecer apenas os seguintes tipos de documentos: artigos científicos originais, revisões, editoriais ou cartas.
Os termos utilizados na pesquisa booleana, para cada teoria, foram os seguintes:
-
- Para a teoria colinérgica: "Alzheimer's disease" (AND) "cholinergic", com ambos os termos estando presentes ou no título ou no resumo dos resultados encontrados.
-
- Para a teoria glutamatérgica: "Alzheimer's disease" (AND) "glutamate", com ambos os termos estando presentes ou no título ou no resumo dos resultados encontrados.
-
- Para a teoria amiloide: "Alzheimer's disease" (AND) "amyloid" (OR) "amyloidogenic", com todos os termos estando presentes ou no título ou no resumo dos resultados encontrados.
-
- Para a teoria oligomérica: "Alzheimer's disease" (AND) "oligomers" (NOT) "tau". Neste caso, exclui-se o termo "tau" da busca, pois a teoria oligomérica refere-se à oligomerização do peptídeo Aβ e não da proteína tau, com ambos os termos estando presentes ou no título ou no resumo dos resultados encontrados.
-
- Para a teoria metálica: "Alzheimer's disease" (AND) "metals". Especificamente, neste caso, optou-se por utilizar um termo mais genérico e não restringir a presença dos termos ao título ou ao resumo, pois esta teoria engloba o estudo de diversos metais, como zinco, cobre e ferro.
-
- Para a teoria diabética: "Alzheimer's disease" (AND) "diabetes", com ambos os termos estando presentes ou no título ou no resumo dos resultados encontrados.
A Figura 4 sumariza os dados obtidos.
Porcentagem de publicações encontradas a respeito das principais hipóteses moleculares para a DA, no período 1980-2014, e as respectivas freqüências de publicação por ano e por teoria
Verifica-se que o número de publicações referentes à hipótese colinérgica estabilizou-se na última década, com uma frequência em torno de 150 publicações por ano. Já as publicações a respeito das outras hipóteses vêm aumentando de maneira relativamente constante nos últimos tempos. Dentre elas, é notável a grande quantidade de publicações a respeito da hipótese amiloide em comparação com as outras hipóteses, com uma frequência aproximadamente 10 vezes maior no ano de 2014. As hipóteses diabética e oligomérica constituem as teorias causais mais recentes, a primeira iniciando o número de publicações na década de 90, e a segunda, a partir dos anos 2000.
PERSPECTIVAS DE TRATAMENTO PARA A DA: ESTADO DA ARTE
Com base nas diferentes hipóteses moleculares da DA, as abordagens dos grupos de pesquisa foram sendo diferenciadas ao longo dos anos. Diferentes tratamentos estão disponíveis atualmente no mercado, tanto aqueles que focam nas hipóteses colinérgica e glutamatérgica, quanto aqueles que auxiliam no controle parcial de diversos sintomas, particularmente agitação, depressão, alucinações e delírios, que são mais frequentes com a progressão da enfermidade. A seguir, estão descritas as terapias atualmente em uso e em desenvolvimento, subdivididas por teoria.
Inibidores da acetilcolinesterase (AChE)
Como visto na hipótese colinérgica, as pessoas que sofrem de DA apresentam níveis baixos de acetilcolina, um importante neurotransmissor. Os inibidores da AChE retardam a degradação metabólica da acetilcolina, otimizando a disponibilidade deste substrato para a comunicação entre as células. Isto auxilia no retardo da progressão da disfunção cognitiva e pode ser eficaz para alguns pacientes nos estágios inicial e intermediário da doença.194194 Anand, P.; Singh, B.; Arch. Pharm. Res.2013 , 36, 375. Existem hoje quatro medicamentos pertencentes a esta classe, aprovados pela Administração Federal de Alimentos e Medicamentos americana (FDA - do inglês, Food and Drug Administration ): tacrina, donepezila, rivastigmina e galantamina. Esses tratamentos foram liberados para sintomas leves a moderados de DA e, dentre eles, somente a donepezila foi aprovada para o tratamento de sintomas severos, em 2006.195195 Cunningham, E. L.; Passmore, A. P.; Maturitas2013 , 76, 260.,196196 Ellul, J.; Archer, N.; Foy, C. M.; Poppe, M.; Boothby, H.; Nicholas, H.; Brown, R. G.; Lovestone, S.; J. Neurol., Neurosurg. Psychiatry2007 , 78, 233.Os inibidores de AChE aprovados para o tratamento da DA estão apresentados naFigura 5.
Tacrina
A tacrina (nome comercial: Cognex® foi aprovada em 1993. Os efeitos colaterais mais comuns são prisão de ventre, diarreia, gases, perda de apetite, dores musculares, náuseas, dor de estômago, nariz entupido, vômitos, perda de peso, e possível hepatotoxicidade. Devido a esses efeitos secundários, a Tacrina não é mais comercializada.194194 Anand, P.; Singh, B.; Arch. Pharm. Res.2013 , 36, 375.,197197 Rodrigues Simões, M. C.; Dias Viegas, F. P.; Moreira, M. S.; de Freitas Silva, M.; Riquiel, M. M.; da Rosa, P. M.; Castelli, M. R.; dos Santos, M. H.; Soares, M. G.; Viegas, C.; Mini Rev. Med. Chem.2014 , 14, 2.
Donepezila
A donepezila (nome comercial: Aricept® foi aprovada em 1996. Os efeitos colaterais mais comuns são diarreia, tonturas, perda de apetite, dores musculares, náuseas, cansaço, problemas para dormir, vômitos e perda de peso. Um estudo de 2005 sugere que esse fármaco pode retardar, levemente, a progressão do MCI associado à DA.198198 Barnes, D. E.; Yaffe, K.; N. Engl. J. Med.2005 , 353, 951. Durante o primeiro ano desse estudo, de um total de três anos, pessoas com MCI tratadas com donepezila tiveram risco reduzido de progressão para DA em comparação com participantes que tomaram vitamina E ou um placebo. No entanto, não houve diferença entre os três grupos após o fim do estudo, exceto para aqueles que apresentavam o gene ApoE4. O efeito da donepezila durou, portanto, de dois a três anos para estes participantes. Estudos anteriores indicam que indivíduos que possuem o gene ApoE4 apresentam maior chance de desenvolver DA do que a população em geral.197197 Rodrigues Simões, M. C.; Dias Viegas, F. P.; Moreira, M. S.; de Freitas Silva, M.; Riquiel, M. M.; da Rosa, P. M.; Castelli, M. R.; dos Santos, M. H.; Soares, M. G.; Viegas, C.; Mini Rev. Med. Chem.2014 , 14, 2.,199199 Michaelson, D. M.; Alzheimer's Dementia2014 , 10, 861.,200200 Molino, I.; Colucci, L.; Fasanaro, A. M.; Traini, E.; Amenta, F.;Sci. World J.2013 , 2013, 925702.
Rivastigmina
A rivastigmina (nome comercial: Exelon® foi aprovada no ano 2000. Este fármaco impede a degradação da acetilcolina, inibindo a acetilcolinesterase e também a butirilcolinesterase (BuChE), uma colinesterase que desempenha papel menor na degradação de acetilcolina no corpo humano. Os efeitos colaterais mais comuns são náuseas, diarreia, vômitos, fraqueza muscular, perda de apetite, perda de peso, tontura, sonolência e dor de estômago. Em 2007, a FDA aprovou o Exelon®Patch, um sistema transdérmico de rivastigmina, para liberar esta medicação por meio de um adesivo para a pele como uma alternativa à cápsula oral.194194 Anand, P.; Singh, B.; Arch. Pharm. Res.2013 , 36, 375.
Galantamina
A galantamina (nome comercial: Razadyne® foi aprovada em 2001. Esse fármaco impede a degradação da acetilcolina e estimula os receptores nicotínicos a liberar maiores quantidades desse neurotransmissor no cérebro. Os efeitos colaterais mais comuns são náuseas, vômitos, diarreia, perda de peso, tontura, dor de cabeça e cansaço.194194 Anand, P.; Singh, B.; Arch. Pharm. Res.2013 , 36, 375.
Atualmente, no Brasil, estão sendo realizadas pesquisas focadas na seleção de plantas com atividade anticolinesterásica para o tratamento da DA, com o intuito de diminuir os custos de produção dos princípios ativos, assim como os efeitos colaterais exibidos pelas drogas sintéticas comercializadas.201201 Trevisan, M. T. S.; Macedo, F. V. V.; Meent, M. v. d.; Rhee, I. K.; Verpoorte, R.; Quim. Nova2003 , 26, 301.,202202 Viegas Junior, C.; Bolzani, V. d. S.; Furlan, M.; Fraga, C. A. M.; Barreiro, E. J.; Quim. Nova2004 , 27, 655.
Antagonistas de receptores de N -metil-d-aspartato (NMDA)
Memantina
A memantina (nome comercial: Namenda®, Figura 6) foi o primeiro fármaco aprovado pela FDA para tratar os sintomas de DA moderada a severa, sendo também o primeiro e, por enquanto, o único representante da classe dos antagonistas de receptores de NMDA. Ela regula a atividade do glutamato, que é liberado em grandes quantidades por células danificadas pela DA e por alguns outros distúrbios neurológicos. Quando o glutamato alcança os receptores do tipo NMDA nas células de superfície, o cálcio flui livremente para dentro da célula, o que pode levar à degeneração celular. A memantina tem a capacidade de evitar esta sequência destrutiva. Por muitos anos, a memantina esteve disponível em alguns países europeus, e está disponível nos EUA desde outubro de 2003. Este fármaco é geralmente bem tolerado pelo paciente, sendo os efeitos colaterais mais comuns cefaleia, constipação, diarreia, tontura e sonolência.2727 Parsons, C. G.; Stöffler, A.; Danysz, W.;Neuropharmacology2007 , 53, 699.,200200 Molino, I.; Colucci, L.; Fasanaro, A. M.; Traini, E.; Amenta, F.;Sci. World J.2013 , 2013, 925702. Em julho de 2010, a Namenda XR®, uma formulação de liberação prolongada do medicamento, foi liberada para uso nos EUA. Os seus efeitos secundários mais comuns são dores de cabeça, diarreia, tonturas e pressão arterial elevada.2727 Parsons, C. G.; Stöffler, A.; Danysz, W.;Neuropharmacology2007 , 53, 699.,200200 Molino, I.; Colucci, L.; Fasanaro, A. M.; Traini, E.; Amenta, F.;Sci. World J.2013 , 2013, 925702.
Agentes quelantes e MPACs
Conforme citado anteriormente, metais fisiológicos como Zn, Cu e Fe estão concentrados ao redor e ligados às placas amiloides em cérebros de pacientes com DA.105105 Deibel, M. A.; Ehmann, W. D.; Markesbery, W. R.; J. Neurol. Sci.1996 , 143, 137. Há evidências de que estes metais interagem com o peptídeo Aβ, catalisando a produção de radicais hidroxila,6060 Hane, F.; Leonenko, Z.; Biomolecules2014 , 4, 101. contribuindo para o estresse oxidativo, gerando fibras amiloides tóxicas, com maior tendência à agregação e resistentes à remoção.111111 Bush, A. I.; Pettingell, W. H.; Multhaup, G.; d Paradis, M.; Vonsattel, J. P.; Gusella, J. F.; Beyreuther, K.; Masters, C. L.; Tanzi, R. E.;Science1994 , 265, 1464.,129129 Bush, A. I.; J. Alzheimer's Dis.2013 , 33, S277.,203203 Finefrock, A. E.; Bush, A. I.; Doraiswamy, P. M.; J. Am. Geriatr. Soc.2003 , 51, 1143.,204204 Bush, A. I.; Trends Neurosci.2003 , 26, 207.
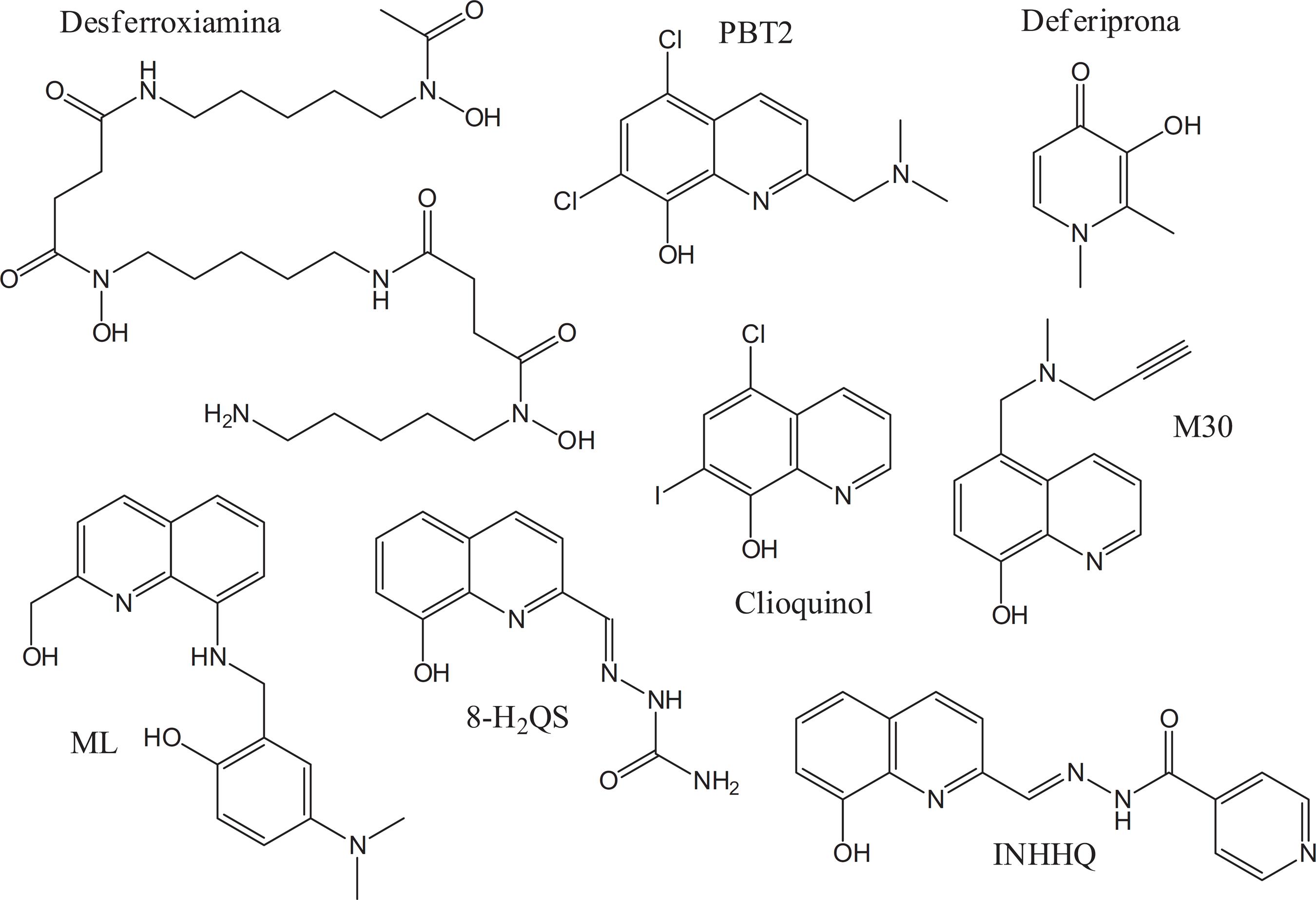
Com base nisso, diversos estudos analisaram a possibilidade do uso de agentes quelantes de metais para o tratamento da DA. Porém, além do fato de grande parte dos quelantes não possuírem a capacidade de ultrapassar a barreira hematoencefálica, a maioria também não mostra especificidade para os íons metálicos complexados com Aβ, levando à quelação indiscriminada de vários biometais e ao aparecimento de graves efeitos adversos devido ao uso prolongado desses compostos.205205 Pardridge, W. M.; Alzheimer's Dementia2009 , 5, 427.,206206 Dedeoglu, A.; Cormier, K.; Payton, S.; Tseitlin, K. A.; Kremsky, J. N.; Lai, L.; Li, X.; Moir, R. D.; Tanzi, R. E.; Bush, A. I.; Kowall, N. W.; Rogers, J. T.; Huang, X.; Exp. Gerontol.2004 , 39, 1641. Os agentes quelantes e MPACs (do inglês, Metal-Protein Attenuating Compounds ) discutidos nesta seção estão apresentados na Figura 7.
A desferroxiamina (DFO) é um agente quelante aprovado pela FDA para o tratamento da hemocromatose, doença caracterizada pelo excesso de ferro, e que, em testes clínicos, demonstrou a capacidade de impedir a progressão da DA devido à quelação e eliminação de Fe3+ do cérebro.207207 Keberle, H.; Ann. N. Y. Acad. Sci.1964 , 119, 758.,208208 Cuajungco, M. P.; Fagét, K. Y.; Huang, X.; Tanzi, R. E.; Bush, A. I.; Ann. N. Y. Acad. Sci.2000 , 920, 292.A DFO também se combina moderadamente com alumínio, zinco e cobre,209209 Richardson, D. R.; Ponka, P.; Am. J. Hematol.1998 , 58, 299.,210210 Olivieri, N. F.; Brittenham, G. M.; Blood1997 , 89, 739. o que pode contribuir para os efeitos atenuadores da DA que esta droga apresenta. Entretanto, por possuir elevado peso molecular e ser bastante hidrofílica, a DFO não consegue atravessar a barreira hematoencefálica, sendo os bons resultados observados devidos à administração subcutânea de longa duração.210210 Olivieri, N. F.; Brittenham, G. M.; Blood1997 , 89, 739.,211211 Kontoghiorghes, G. J.; Analyst1995 , 120, 845. Da mesma forma, a deferiprona é um agente quelante de ferro e alumínio aprovado para uso terapêutico na Europa, mas não nos Estados Unidos.212212 Reichard, P.; Annu. Rev. Biochem.1988 , 57, 349. Este composto possui efeitos quelantes moderados, mas seu tamanho pequeno e lipofilicidade a tornam ligeiramente tóxica quando administrada oralmente. Entretanto, a facilidade que a deferiprona possui para atravessar a barreira hematoencefálica permite que ela remova rapidamente o ferro de pools intracelulares, sendo interessante no contexto da DA.120120 Bonda, D. J.; Lee, H. G.; Blair, J. A.; Zhu, X.; Perry, G.; Smith, M. A.; Metallomics2011 , 3, 267.
Algumas pesquisas têm sido realizadas com drogas híbridas, ou seja, drogas que apresentam diferentes funções concomitantes no organismo, atacando ao mesmo tempo duas ou mais facetas da DA. Destacam-se neste contexto drogas com função dupla de quelação e de interação com Aβ, AChE, β secretase, ou monoamina oxidase, ou com ação antioxidante, como reportado em uma recente revisão de Huang e colaboradores sobre agentes quelantes com potencial atividade no tratamento da DA.213213 Huang, W.; Wei, W.; Shen, Z.; RSC Adv.2014 , 4, 52088. Nesse cenário, duas moléculas específicas são de especial intresse: M30214214 Kupershmidt, L.; Amit, T.; Bar-Am, O.; Weinreb, O.; Youdim, M. B.;Mol. Neurobiol.2012 , 46, 217.,215215 Zheng, H.; Gal, S.; Weiner, L. M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M. B.; J. Neurochem.2005 , 95, 68.e ML.216216 Lee, S.; Zheng, X.; Krishnamoorthy, J.; Savelieff, M. G.; Park, H. M.; Brender, J. R.; Kim, J. H.; Derrick, J. S.; Kochi, A.; Lee, H. J.; Kim, C.; Ramamoorthy, A.; Bowers, M. T.; Lim, M. H.; J. Am. Chem. Soc.2014 , 136, 299.
O M30, 5-[N -metil-N-propargilaminometil]-8-hidroxiquinolina tem mostrado a capacidade de reduzir o acúmulo de Aβ e a fosforilação da proteína tau in vitro, e diminuir as deficiências mnemônicas nos modelos testados in vivo, direcionando a pesquisa para o estudo de outras moléculas derivadas das benzilideinoindanonas.213213 Huang, W.; Wei, W.; Shen, Z.; RSC Adv.2014 , 4, 52088.
214 Kupershmidt, L.; Amit, T.; Bar-Am, O.; Weinreb, O.; Youdim, M. B.;Mol. Neurobiol.2012 , 46, 217.-215215 Zheng, H.; Gal, S.; Weiner, L. M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M. B.; J. Neurochem.2005 , 95, 68. Já o ML, acrônimo de Multifunctional Ligand, ligante multifuncional, é uma potencial droga híbrida obtida por meio da integração racional de elementos estruturais com o objetivo de atuar de quatro formas diferentes: desagregação de Aβ, quelação de metais, regulação das espécies reativas de oxigênio e atividade antioxidante. Este composto, solúvel em água, possui a capacidade de interagir com diferentes espécies, tanto solúveis quanto insolúveis, de Aβ, além de ter potencial para atravessar a barreira hematoencefálica.216216 Lee, S.; Zheng, X.; Krishnamoorthy, J.; Savelieff, M. G.; Park, H. M.; Brender, J. R.; Kim, J. H.; Derrick, J. S.; Kochi, A.; Lee, H. J.; Kim, C.; Ramamoorthy, A.; Bowers, M. T.; Lim, M. H.; J. Am. Chem. Soc.2014 , 136, 299.
Por outro lado, os compostos atenuadores da interação metal-proteína (MPACs) diferem conceitualmente de agentes quelantes tradicionais, pois possuem afinidade moderada por íons metálicos. Assim, ao invés de se ligarem e removerem sistematicamente os íons dos tecidos afetados, eles inibem apenas as interações metálicas anormais, resultando em efeitos sutis na homeostase dos metais, o que levaria à inibição da oligomerização de Aβ induzida por Zn2+ e Cu2+, favorecendo a solubilização e remoção do peptídeo Aβ, dificultando também as reações de oxirredução que levam à geração de radicais livres. Consequentemente, os MPACs representam uma estratégia terapêutica viável para retardar ou mesmo prevenir o progresso da demência associada à DA.123123 Barnham, K. J.; Masters, C. L.; Bush, A. I.; Nat. Rev. Drug Discov.2004 , 3, 205.,213213 Huang, W.; Wei, W.; Shen, Z.; RSC Adv.2014 , 4, 52088.,217217 Sampson, E. L.; Jenagaratnam, L.; McShane, R.; Cochrane Database of Systematic Reviews2012 , 5, CD005380.,218218 Cherny, R. A.; Atwood, C. S.; Xilinas, M. E.; Gray, D. N.; Jones, W. D.; McLean, C. A.; Barnham, K. J.; Volitakis, I.; Fraser, F. W.; Kim, Y.; Huang, X.; Goldstein, L. E.; Moir, R. D.; Lim, J. T.; Beyreuther, K.; Zheng, H.; Tanzi, R. E.; Masters, C. L.; Bush, A. I.; Neuron2001 , 30, 665.
O clioquinol (CQ, 5-cloro-7-iodo-8-hidroxiquinolina) pertence à classe das 8-hidroxiquinolinas, sendo o primeiro MPAC a ser proposto como possível fármaco para a terapia da DA.219219 Melov, S.; Trend Neurosci.2002 , 25, 121.
220 Doraiswamy, P. M.; Finefrock, A. E.; Lancet Neurol.2004 , 3, 431.
221 Bareggi, S. R.; Cornelli, U.; CNS Neurosci. Ther.2012 , 18, 41.-222222 Regland, B.; Lehmann, W.; Abedini, I.; Blennow, K.; Jonsson, M.; Karlsson, I.; Sjögren, M.; Wallin, A.; Xilinas, M.; Gottfries, C. G.;Dementia Geriatr. Cognit. Disord.2001 , 12, 408. É um quelante pequeno, lipofílico e biodisponível, com alta seletividade por íons Zn2+ e Cu2+ e capacidade de atravessar a barreira hematoencefálica em cobaias.218218 Cherny, R. A.; Atwood, C. S.; Xilinas, M. E.; Gray, D. N.; Jones, W. D.; McLean, C. A.; Barnham, K. J.; Volitakis, I.; Fraser, F. W.; Kim, Y.; Huang, X.; Goldstein, L. E.; Moir, R. D.; Lim, J. T.; Beyreuther, K.; Zheng, H.; Tanzi, R. E.; Masters, C. L.; Bush, A. I.; Neuron2001 , 30, 665.,223223 Budimir, A.; Acta Pharm.2011 , 61, 1. Embora usado originalmente como uma substância anti-amébica, o CQ mostrou também a capacidade de diminuir a formação de placas β-amiloides em modelos transgênicos de ratos para a DA, atribuída à remoção de metais das placas.224224 Mancino, A. M.; Hindo, S. S.; Kochi, A.; Lim, M. H.; Inorg. Chem.2009 , 48, 9596. Infelizmente, porém, pesquisas apontaram como principal efeito colateral uma elevada correlação com neuropatia mielo-óptica subaguda,220220 Doraiswamy, P. M.; Finefrock, A. E.; Lancet Neurol.2004 , 3, 431.,223223 Budimir, A.; Acta Pharm.2011 , 61, 1.,225225 Levine, H.; Dingb, Q.; Walkerc, J. A.; Vossc, R. A.; Augelli-Szafrand, C. E.; Neurosci. Lett.2009 , 465, 99.,226226 Adlard, P. A.; Cherny, R. A.; Finkelstein, D. I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J. P.; Perez, K.; Laughton, K.; Li, Q. X.; Charman, S. A.; Nicolazzo, J. A.; Wilkins, S.; Deleva, K.; Lynch, T.; Kok, G.; Ritchie, C. W.; Tanzi, R. E.; Cappai, R.; Masters, C. L.; Barnham, K. J.; Bush, A. I.; Neuron2008 , 59, 43. o que estimulou a busca por novos análogos menos tóxicos.220220 Doraiswamy, P. M.; Finefrock, A. E.; Lancet Neurol.2004 , 3, 431.,226226 Adlard, P. A.; Cherny, R. A.; Finkelstein, D. I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J. P.; Perez, K.; Laughton, K.; Li, Q. X.; Charman, S. A.; Nicolazzo, J. A.; Wilkins, S.; Deleva, K.; Lynch, T.; Kok, G.; Ritchie, C. W.; Tanzi, R. E.; Cappai, R.; Masters, C. L.; Barnham, K. J.; Bush, A. I.; Neuron2008 , 59, 43.Com isso, uma nova geração de compostos derivados do clioquinol tem sido estudada por diferentes grupos de pesquisa. O ligante PBT2 demonstrou potencial terapêutico para DA em modelos murinos,226226 Adlard, P. A.; Cherny, R. A.; Finkelstein, D. I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J. P.; Perez, K.; Laughton, K.; Li, Q. X.; Charman, S. A.; Nicolazzo, J. A.; Wilkins, S.; Deleva, K.; Lynch, T.; Kok, G.; Ritchie, C. W.; Tanzi, R. E.; Cappai, R.; Masters, C. L.; Barnham, K. J.; Bush, A. I.; Neuron2008 , 59, 43.,227227 Bush, A. I.; J. Alzheimer's Dis.2008 , 15, 223. assim como em fase 2 de testes clínicos.228228 Faux, N. G.; Ritchie, C. W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; Ingelsson, M.; Masters, C. L.; Tanzi, R. E.; Cummings, J. L.; Herd, C. M.; Bush, A. I.;J. Alzheimer's Dis.2010 , 20, 509.,229229 Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C. L.; Targum, S.; Bush, A. I.; Murdoch, R.; Wilson, J.; Ritchie, C. W.; Lancet Neurol.2008 , 7, 779. Este composto reduz a agregação de Aβ, limita a toxicidade dos oligômeros e redistribui íons metálicos nos neurônios.230230 Crouch, P. J.; Savva, M. S.; Hung, L. W.; Donnelly, P. S.; Mot, A. I.; Parker, S. J.; Greenough, M. A.; Volitakis, I.; Adlard, P. A.; Cherny, R. A.; Masters, C. L.; Bush, A. I.; Barnham, K. J.; White, A. R.; J. Neurochem.2011 , 119, 220.,231231 Crouch, P. J.; Barnham, K. J.; Acc. Chem. Res.2012 , 45, 1604.
No Brasil, esforços também vêm sendo realizados neste sentido. Por exemplo, um grupo de pesquisa da Universidade Federal de Minas Gerais, em parceria com a Simon Fraser University, no Canadá, publicou recentemente um estudo que propõe duas diferentes substâncias derivadas do 8-hidroxiquinolina-2-carboxaldeído como potenciais MPACs para o tratamento da DA. Os resultados sugerem que os compostos apresentam a capacidade de atravessar a barreira hematoencefálica, de se ligar ao cobre em condições fisiológicas e de diminuir a agregação de Aβ na presença de Cu2+. A mais ativa dessas substâncias é 8-H2QS, uma semicarbazona. O estudo também indica que esses compostos exibem atividade antioxidante similar à da vitamina E.232232 Gomes, L. M.; Vieira, R. P.; Jones, M. R.; Wang, M. C.; Dyrager, C.; Souza-Fagundes, E. M.; Da Silva, J. G.; Storr, T.; Beraldo, H.; J. Inorg. Biochem.2014 , 139, 106.
Outro estudo, desta vez conduzido na Pontifícia Universidade Católica do Rio de Janeiro, aponta para resultados promissores obtidos com um outro ligante derivado da 8-hidroxiquinolina, o INHHQ,233233 de Freitas, L. V.; da Silva, C. C.; Ellena, J.; Costa, L. A.; Rey, N. A.; Spectrochim. Acta, Part A2013 , 116, 41. alvo de uma solicitação de patente brasileira (BR 10 2013 033006 0) e internacional (PCT/BR2014/000186). Um estudo realizado in vitro234234 Hauser-Davis, R. A.; de Freitas, L. V.; Cukierman, D. S.; Cruz, W. S.; Miotto, M. C.; Landeira-Fernandez, J.; Valiente-Gabioud, A. A.; Fernández, C. O.; Rey, N. A.; Metallomics2015 , 7, 743. demonstrou que esta hidrazona é capaz de bloquear as interações de Zn2+ e Cu2+ com o peptídeo Aβ. Análises farmacológicas in silico também demonstraram que o composto é neutro em pH fisiológico, e que é capaz de ultrapassar a barreira hematoencefálica. O parâmetro chamado DrugScore, que estima a probabilidade de um composto se tornar um fármaco comercial, foi calculado em cerca de 70% para o INHHQ. Além disso, testes de toxicidade aguda em ratos sadios foram conduzidos, e não foram observadas diferenças entre os níveis de proteínas biomarcadoras de estresse oxidativo e concentrações de biometais no cérebro de animais injetados com o INHHQ e o grupo controle, demonstrando que o INHHQ não age como um agente quelante tradicional, e sim como um promissor MPAC no contexto do tratamento da DA. Apesar da presença do grupo 8-hidroxiquinolina na estrutura de INHHQ, o que é uma constante em relação à maioria dos MPACs, é importante ressaltar que a coordenação dos biometais cobre e zinco se dá, nesse composto, por meio do sistema hidrazônico, abrindo novas perspectivas em relação ao desenvolvimento de promissores atenuadores da interação metal-proteína.
Fármacos contra o diabetes
Ao longo dos últimos anos, diferentes estudos propuseram, com base na correlação entre o diabetes e a DA, o uso de fármacos contra o diabetes para o tratamento da progressão da doença.169169 Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L.;Neurology2008 , 71, 1065.,185185 de la Monte, S. M.; Tong, M.; Lester-Coll, N.; Plater, M.; Wands, J. R.; J. Alzheimer's Dis.2006 , 10, 89.,187187 Lester-Coll, N.; Rivera, E. J.; Soscia, S. J.; Doiron, K.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2006 , 9, 13.
188 Craft, S.; Watson, G. S.; Lancet Neurol.2004 , 3, 169.
189 Reger, M. A.; Watson, G. S.; Green, P. S.; Wilkinson, C. W.; Baker, L. D.; Cholerton, B.; Fishel, M. A.; Plymate, S. R.; Breitner, J. C.; DeGroodt, W.; Mehta, P.; Craft, S.; Neurology2008 , 70, 440.-190190 Park, C. R.; Seeley, R. J.; Craft, S.; Woods, S. C.;Physiol. Behav.2000 , 68, 509.
Em 2011, um estudo de revisão levantou dados de ensaios clínicos de terapias farmacológicas, em uso e ainda em fase experimental, para tratar o diabetesmellitus tipo 2. A análise abrangeu estudos a respeito de antagonistas do receptor ativado por proliferadores de peroxissoma γ, que se encontra aumentado no cérebro de pacientes com DA, e sugeriu, com base nos estudos avaliados, que o uso desses antagonistas permite conferir algum benefício terapêutico para paciente em fases iniciais de DA. No estudo, foram avaliados os efeitos da administração de insulina intranasal, aproveitando a capacidade da insulina (Figura 8) atingir rapidamente o cérebro por meio de transporte através do canal olfativo, trigêmeo e via axonal, revelando que a eficácia desta exposição é diretamente dependente do genótipo ApoE4 dos pacientes avaliados.235235 Akter, K.; Lanza, E. A.; Martin, S. A.; Myronyuk, N.; Rua, M.; Raffa, R. B.; Br. J. Clin. Pharmacol.2011 , 71, 365.
Uma análise mais recente propôs, com base nos dados de correlação molecular entre síndromes inflamatórias, sinalização desregulamentada da insulina e disfunção mitocondrial em DA e diabetes, o desenvolvimento de novas estratégias terapêuticas com base em agentes antidiabéticos e/ou anti-inflamatórios, apontando, porém, para a necessidade de mais estudos voltados para desvendar os mecanismos implicados na inflamação do cérebro e sinalização desregulada da insulina na DA.186186 De Felice, F. G.; Lourenco, M. V.; Ferreira, S. T.;Alzheimer's Dementia2014 , 10, S26.
Terapia de suporte: fármacos para o controle da depressão
Com a progressão da DA, cerca de 60% dos pacientes passam a apresentar sintomas neuropsiquiátricos, como depressão, agitação e sintomas psicóticos como pensamentos paranoicos, delírios e/ou alucinações.236236 Lyketsos, C. G.; Steinberg, M.; Tschanz, J. T.; Norton, M. C.; Steffens, D. C.; Breitner, J. C.; Am. J. Psychiatry2000 , 157, 708. Os sintomas podem ter uma origem médica subjacente, como a interação medicamentosa ou dor física. O uso de antidepressivos é comum na demência, com uma prevalência de 43,2% relatada em um recente estudo dinamarquês.236236 Lyketsos, C. G.; Steinberg, M.; Tschanz, J. T.; Norton, M. C.; Steffens, D. C.; Breitner, J. C.; Am. J. Psychiatry2000 , 157, 708.,237237 Kessing, L. V.; Harhoff, M.; Andersen, P. K.; International Psychogeriatrics2007 , 19, 902. Os fármacos para o controle da depressão discutidos nesta seção são apresentados na Figura 9.
Estrutura dos principais fármacos para o controle da depressão utilizados no tratamento da DA
Diversos antidepressivos têm sido estudados no contexto da DA, porém a interpretação dos efeitos clínicos do tratamento é controversa devido à ambiguidade dos resultados. Por exemplo, um estudo a respeito de um inibidor seletivo da recaptação de serotonina (SSRI - do inglês, Selective Serotonin Re-uptake Inhibitor ), o cloridrato de sertralina, indicou resultados positivos, em contraste com outros estudos a respeito do mesmo fármaco, e ainda outros dois antidepressivos: fluoxetina e citalopram.238238 Reifler, B. V.; Teri, L.; Raskind, M.; Veith, R.; Barnes, R.; White, E.; McLean, P.; Am. J. Psychiatry1989 , 146, 45.
239 Nyth, A. L.; Gottfries, C. G.; Br. J. Psychiatry1990 , 157, 894.-240240 Magai, C.; Kennedy, G.; Cohen, C. I.; Gomberg, D.; American Journal of Geriatric Psychiatry2000 , 8, 66. Na Europa, existe um consenso sobre a indicação do tratamento baseado em SSRIs,241241 Caltagirone, C.; Bianchetti, A.; Di Luca, M.; Mecocci, P.; Padovani, A.; Pirfo, E.; Scapicchio, P.; Senin, U.; Trabucchi, M.; Musicco, M.;Drugs Aging2005 , 22, 1. apesar de esta recomendação terapêutica não ser suportada por evidências conclusivas, pois alguns ensaios clínicos mostraram efeitos benéficos no estado de humor dos pacientes,239239 Nyth, A. L.; Gottfries, C. G.; Br. J. Psychiatry1990 , 157, 894.,242242 Lyketsos, C. G.; DelCampo, L.; Steinberg, M.; Miles, Q.; Steele, C. D.; Munro, C.; Baker, A. S.; Sheppard, J. M.; Frangakis, C.; Brandt, J.; Rabins, P. V.; Arch. Gen. Psychiatry2003 , 60, 737. dado não confirmado em outro estudo independente.243243 Petracca, G. M.; Chemerinski, E.; Starkstein, S. E.;International Psychogeriatrics2001 , 13, 233. Um ensaio clínico randomizado sugeriu que o tratamento com antidepressivos melhora a função cognitiva em indivíduos deprimidos com demência,244244 Nyth, A. L.; Gottfries, C. G.; Lyby, K.; Smedegaard-Andersen, L.; Gylding-Sabroe, J.; Kristensen, M.; Refsum, H. E.; Ofsti, E.; Eriksson, S.; Syversen, S.; Acta Psychiatr. Scand.1992 , 86, 138. e um estudo mais recente mostrou efeitos positivos destes agentes sobre a atividade funcional, mas não confirmou a melhoria da atividade cognitiva dos pacientes.245245 Munro, C. A.; Brandt, J.; Sheppard, J. M.; Steele, C. D.; Samus, Q. M.; Steinberg, M.; Rabins, P. V.; Lyketsos, C. G.; American Journal of Geriatric Psychiatry2004 , 12, 491. Todos esses estudos trazem evidências muito limitadas, devido às pequenas dimensões da população amostrada e ao curto prazo de observação.
Atualmente, outros estudos estão sendo conduzidos para determinar se os antidepressivos podem ser utilizados como agentes preventivos da doença, e não somente prescritos em estágios avançados, com base na hipótese destes compostos tratarem não apenas os sintomas neuropsiquiátricos, mas também de serem capazes de reduzir a produção de Aβ.246246 Sheline, Y. I.; West, T.; Yarasheski, K.; Swarm, R.; Jasielec, M. S.; Fisher, J. R.; Ficker, W. D.; Yan, P.; Xiong, C.; Frederiksen, C.; Grzelak, M. V.; Chott, R.; Bateman, R. J.; Morris, J. C.; Mintun, M. A.; Lee, J. M.; Cirrito, J. R.; Sci. Transl. Med.2014 , 6, 236re4.
Antioxidantes
A Figura 10 apresenta as estruturas dos agentes antioxidantes discutidos nesta seção.
A vitamina E, também conhecida como α-tocoferol, é um antioxidante e tem sido prescrita para tratar os sintomas cognitivos da DA.198198 Barnes, D. E.; Yaffe, K.; N. Engl. J. Med.2005 , 353, 951. Seu uso nesta doença foi baseado principalmente em um estudo realizado em 1997, com o qual se demonstrou que altas doses deste composto ingeridas durante diversos meses causam a desaceleração da perda da capacidade de realizar atividades diárias.247247 Adelman, A.; J. Fam. Pract.1997 , 45, 98. Porém, nos anos seguintes a este estudo, foram geradas evidências que apontam ao fato de altas doses de vitamina E provocarem aumento do risco de morte, especialmente em pacientes com doença arterial coronariana.248248 Spencer, A. P.; Carson, D. S.; Crouch, M. A.; Arch. Intern. Med.1999 , 159, 1313. Recentemente, em 2014, os resultados de um estudo indicaram que os indivíduos com DA de leve a moderada que receberam altas doses de vitamina E tiveram uma taxa 19% mais lenta de declínio funcional do que os voluntários que receberam um placebo.249249 Dysken, M. W.; Sano, M.; Asthana, S.; Vertrees, J. E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G. D.; McCarten, J. R.; Malphurs, J.; Prieto, S.; Chen, P.; Loreck, D. J.; Trapp, G.; Bakshi, R. S.; Mintzer, J. E.; Heidebrink, J. L.; Vidal-Cardona, A.; Arroyo, L. M.; Cruz, A. R.; Zachariah, S.; Kowall, N. W.; Chopra, M. P.; Craft, S.; Thielke, S.; Turvey, C. L.; Woodman, C.; Monnell, K. A.; Gordon, K.; Tomaska, J.; Segal, Y.; Peduzzi, P. N.; Guarino, P. D.; JAMA2014 , 311, 33. Os participantes do estudo foram acompanhados por pouco mais de dois anos. Os pacientes que receberam a vitamina E em conjunto com memantina não mostraram o mesmo benefício que os participantes que receberam somente a vitamina E, e nenhum dos quatro grupos de estudo (placebo, vitamina E, memantina, vitamina E em conjunto com memantina) mostrou melhoras cognitivas.
Por outro lado, a CoQ é um antioxidante natural que auxilia na proteção das mitocôndrias e membranas lipídicas contra espécies reativas de oxigênio formadas durante o metabolismo oxidativo, ao agir como co-fator na cadeia transportadora de elétrons.250250 Beal, M. F.; J. Bioenerg. Biomembr.2004 , 36, 381. CoQ10(2,3-dimetoxi-5-metil-6-decaprenilbenzoquinona), também conhecida como ubiquinona, é a forma predominante desta co-enzima. Diversos estudos in vivo avaliaram os efeitos da sua administração em cobaias, obtendo resultados interessantes no contexto da DA. Por exemplo, a administração desta co-enzima em ratos transgênicos idosos levou a uma diminuição nos níveis corticais de Aβ1-42 e à redução dos marcadores de estresse oxidativo,251251 Yang, X.; Yang, Y.; Li, G.; Wang, J.; Yang, E. S.; J. Mol. Neurosci.2008 , 34, 165. enquanto um estudo com camundongos idosos mostrou diminuição dos níveis de carbonilas cerebrais e outros marcadores de estresse oxidativo.252252 Wadsworth, T. L.; Bishop, J. A.; Pappu, A. S.; Woltjer, R. L.; Quinn, J. F.; J. Alzheimer's Dis.2008 , 14, 225. Os efeitos da formulação hidrossolúvel da CoQ10(WS-CoQ10) foram investigados em uma linhagem celular de fibroblastos modelo da DA, e os resultados indicam que a exposição a WS-CoQ10 diminuiu a produção de espécies reativas de oxigênio, aumentou as duplicações da população celular e adiou a síndrome de senescência prematura induzida pelo stress oxidativo.253253 Ma, D.; Stokes, K.; Mahngar, K.; Domazet-Damjanov, D.; Sikorska, M.; Pandey, S.; Mitochondrion2014 , 17, 106. Já estudos em seres humanos examinaram os efeitos de altas doses de CoQ, não mostrando toxicidade ou intolerância em pacientes com DA, porém sem melhora dos índices de estresse oxidativo ou de neurodegeneração.254254 Galasko, D. R.; Peskind, E.; Clark, C. M.; Quinn, J. F.; Ringman, J. M.; Jicha, G. A.; Cotman, C.; Cottrell, B.; Montine, T. J.; Thomas, R. G.; Aisen, P.; Study, A. s. D. C.; Arch. Neurol.2012 , 69, 836. Estes resultados não têm suportado a continuação do desenvolvimento de ensaios clínicos com a CoQ no contexto da DA.
O ácido α-lipóico (ALA) é uma co-enzima mitocondrial com ações antioxidantes diretas e indiretas.255255 Smith, A. R.; Shenvi, S. V.; Widlansky, M.; Suh, J. H.; Hagen, T. M.; Curr. Med. Chem.2004 , 11, 1135. Em um estudo realizado com este composto em 2008, cães idosos receberam uma dieta rica em suplementos antioxidantes, incluindo ALA, apresentando melhor desempenho cognitivo, diminuição da carga amiloide, assim como redução dos danos oxidativos.256256 Cotman, C. W.; Head, E.; J. Alzheimer's Dis.2008 , 15, 685. Estudos a longo prazo da suplementação dietética de ALA em ratos transgênicos relataram melhoria dos animais em testes de memória e diminuição do marcadores de oxidação, sem mudanças na patologia amiloide.257257 Quinn, J. F.; Bussiere, J. R.; Hammond, R. S.; Montine, T. J.; Henson, E.; Jones, R. E.; Stackman, R. W.; Neurobiol. Aging2007 , 28, 213.,258258 Siedlak, S. L.; Casadesus, G.; Webber, K. M.; Pappolla, M. A.; Atwood, C. S.; Smith, M. A.; Perry, G.; Free Radic. Res.2009 , 43, 156. Já um estudo de coorte com pacientes nos estágios iniciais da DA demonstrou estabilização cognitiva dos indivíduos que receberam doses de ALA ao longo de 48 meses, em comparação com aqueles que não as receberam.259259 Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G.;J. Neural Transm., Suppl.2007 , 189. Em outro estudo com humanos, foram testadas doses de ALA em conjunto com as vitaminas E e C, e os resultados demonstraram um aumento do declínio do estado mental dos pacientes, porém sem correlação com as alterações nas medidas dos biomarcadores do fluido cerebrospinal relacionados com Aβ e com a proteína tau, sugerindo que as alterações cognitivas não seriam devidas ao agravamento da patologia relacionada com a DA.254254 Galasko, D. R.; Peskind, E.; Clark, C. M.; Quinn, J. F.; Ringman, J. M.; Jicha, G. A.; Cotman, C.; Cottrell, B.; Montine, T. J.; Thomas, R. G.; Aisen, P.; Study, A. s. D. C.; Arch. Neurol.2012 , 69, 836.
Polifenóis selecionados a partir de plantas de uso alimentar comum já foram confirmados como agentes neuroprotetores,201201 Trevisan, M. T. S.; Macedo, F. V. V.; Meent, M. v. d.; Rhee, I. K.; Verpoorte, R.; Quim. Nova2003 , 26, 301.,202202 Viegas Junior, C.; Bolzani, V. d. S.; Furlan, M.; Fraga, C. A. M.; Barreiro, E. J.; Quim. Nova2004 , 27, 655. inclusive reduzindo a agregação do peptídeo Aβ.260260 Smid, S. D.; Maag, J. L.; Musgrave, I. F.; Food Funct.2012 , 3, 1242. Em particular, o trans-3,5,4'-triidroxiestilbeno, mais conhecido como resveratrol, encontrado predominantemente nas uvas utilizadas para a produção de vinho tinto e em cereais, chá e amendoins, foi testado em diferentes modelos da doença, tantoin vivo quanto in vitro, apresentando resultados positivos como neuroprotetor e inibidor da agregação de Aβ.261261 Richard, T.; Pawlus, A. D.; Iglésias, M. L.; Pedrot, E.; Waffo-Teguo, P.; Mérillon, J. M.; Monti, J. P.; Ann. N. Y. Acad. Sci.2011 , 1215, 103.
262 Regitz, C.; Fitzenberger, E.; Mahn, F. L.; Dußling, L. M.; Wenzel, U.; Eur. J. Nutr.2015 ,-263263 Rege, S. D.; Geetha, T.; Broderick, T. L.; Babu, J. R.;Curr. Alzheimer Res.2015 , 12, 147. Existem também evidências sugerindo que este composto apresenta efeitos anti-diabéticos, anti-inflamatórios e anticancerígenos.263263 Rege, S. D.; Geetha, T.; Broderick, T. L.; Babu, J. R.;Curr. Alzheimer Res.2015 , 12, 147.,264264 Bertelli, A. A.; Ferrara, F.; Diana, G.; Fulgenzi, A.; Corsi, M.; Ponti, W.; Ferrero, M. E.; Bertelli, A.; Int. J. Tissue React.1999 , 21, 93. Outros polifenóis com capacidade de eliminação de radicais livres e de neuroproteção são os da classe dos flavonóides.265265 Mercer, L. D.; Kelly, B. L.; Horne, M. K.; Beart, P. M.;Biochem. Pharmacol.2005 , 69, 339.Estudos com um destes compostos, a quercetina,266266 Selvaraj, K.; Chowdhury, R.; Bhattacharjee, C.; Int. J. Pharm. Pharm. Sci.2013 , 5, demonstraram que ela contribui significativamente na proteção das células neuronais contra a neurotixicidade causada pelo estresse oxidativo,267267 Heo, H. J.; Lee, C. Y.; J. Agric. Food Chem.2004 , 52, 7514. amenizando os efeitos deletérios do peptídeo Aβ.268268 Ansari, M. A.; Abdul, H. M.; Joshi, G.; Opii, W. O.; Butterfield, D. A.; J. Nutr. Biochem.2009 , 20, 269.
Outras abordagens terapêuticas: inibidores de secretases, anticorpos contra Aβ e compostos capazes de dissolver emaranhados neurofibrilares
A Figura 11 apresenta as estruturas dos compostos abordados nesta seção.
A abordagem mais óbvia para limitar o acúmulo de Aβ nos tecidos seria diminuir a produção deste peptídeo. Teoricamente, este resultado poderia ser obtido reduzindo-se a atividade das β (BACE) e γ secretases.269269 Dobrowolska, J. A.; Michener, M. S.; Wu, G.; Patterson, B. W.; Chott, R.; Ovod, V.; Pyatkivskyy, Y.; Wildsmith, K. R.; Kasten, T.; Mathers, P.; Dancho, M.; Lennox, C.; Smith, B. E.; Gilberto, D.; McLoughlin, D.; Holder, D. J.; Stamford, A. W.; Yarasheski, K. E.; Kennedy, M. E.; Savage, M. J.; Bateman, R. J.; J. Neurosci.2014 , 34, 8336. Na prática, o desenvolvimento de substâncias anti-BACE é prejudicado pela possibilidade de que os inibidores possam agir simultaneamente sobre diferentes substratos, como no caso do heparan sulfato e da heparina, inibidores in vitro de BACE-1,270270 Jacobsen, H.; Ozmen, L.; Caruso, A.; Narquizian, R.; Hilpert, H.; Jacobsen, B.; Terwel, D.; Tanghe, A.; Bohrmann, B.; J. Neurosci.2014 , 34, 11621.
271 Patey, S. J.; Edwards, E. A.; Yates, E. A.; Turnbull, J. E.;J. Med. Chem.2006 , 49, 6129.-272272 Scholefield, Z.; Yates, E. A.; Wayne, G.; Amour, A.; McDowell, W.; Turnbull, J. E.; J. Cell. Biol.2003 , 163, 97. utilizados como anticoagulantes e, por esse motivo, pouco adequados para tratamentos de longa duração. Em 2012, dados dos testes de fase I do inibidor MK-8931 reportaram uma diminuição de Aβ superior a 90% no fluido cérebro-espinhal dos pacientes testados, estimulando a pesquisa a prosseguir para as fases II e III para verificar os efeitos secundários da inibição do BACE-1, alvo da droga, a longo prazo.273273 Menting, K. W.; Claassen, J. A.; Front. Aging Neurosci.2014 , 6, 165.
Vale a pena ressaltar que a inibição das secretases impede a formação de novos peptídeos, porém não influencia os peptídeos já existentes. Sendo assim, este tipo de inibição pode ser considerada uma potencial forma de prevenção da DA em vez de um real tratamento terapêutico.
Alternativamente, terapias baseadas no uso de anticorpos contra Aβ têm sido consideradas. Nas condições experimentais, a produção de Aβ1-42 é limitada também por alguns anti-inflamatórios não-esteroides, como ibuprofeno, indometacina e sulindac,274274 Imbimbo, B. P.; Giardina, G. A.; Curr. Top. Med. Chem.2011 , 11, 1555. que favorecem a transformação de Aβ1-42 em um peptídeo menor e não-neurotóxico devido à ação dessas substâncias sobre as secretases, independentemente de seus efeitos anti-inflamatórios. O interesse por estas moléculas iniciou-se com a observação da diminuição do risco de demência em indivíduos que faziam uso desses compostos de maneira regular. Contudo, o uso controlado de anti-inflamatórios não tem se mostrado capaz de limitar a deterioração cognitiva.274274 Imbimbo, B. P.; Giardina, G. A.; Curr. Top. Med. Chem.2011 , 11, 1555. Para impedir que o Aβ seja produzido em excesso e, portanto, se acumule, um estudo está sendo desenvolvido para incrementar a atividade da neprilisina e de outras enzimas proteolíticas capazes de atacar e digerir o peptídeo. A pesquisa nessa direção é conduzida não só em termos de ativação farmacológica, mas também em termos de terapia gênica para aumentar a disponibilidade das enzimas.275275 Plucińska, K.; Crouch, B.; Koss, D.; Robinson, L.; Siebrecht, M.; Riedel, G.; Platt, B.; J. Neurosci.2014 , 34, 10710.
O azul de metileno, antes conhecido apenas como um corante celular e tecidual com leve capacidade bactericida, demonstrou ser capaz de dissolver emaranhados neurofibrilares,276276 Wischik, C. M.; Edwards, P. C.; Lai, R. Y.; Roth, M.; Harrington, C. R.; Proc. Natl. Acad. Sci. U. S. A.1996 , 93, 11213. reduzir níveis de proteína tau e causar aumento da atividade do citocromo-c oxidase e melhoria da memória espacial de cobaias transgênicas.276276 Wischik, C. M.; Edwards, P. C.; Lai, R. Y.; Roth, M.; Harrington, C. R.; Proc. Natl. Acad. Sci. U. S. A.1996 , 93, 11213.
277 Callaway, N. L.; Riha, P. D.; Bruchey, A. K.; Munshi, Z.; Gonzalez-Lima, F.; Pharmacol. Biochem. Behav.2004 , 77, 175.
278 Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M.; J. Biol. Chem.2005 , 280, 7614.
279 Congdon, E. E.; Wu, J. W.; Myeku, N.; Figueroa, Y. H.; Herman, M.; Marinec, P. S.; Gestwicki, J. E.; Dickey, C. A.; Yu, W. H.; Duff, K. E.;Autophagy2012 , 8, 609.-280280 Jinwal, U. K.; Miyata, Y.; Koren, J.; Jones, J. R.; Trotter, J. H.; Chang, L.; O'Leary, J.; Morgan, D.; Lee, D. C.; Shults, C. L.; Rousaki, A.; Weeber, E. J.; Zuiderweg, E. R.; Gestwicki, J. E.; Dickey, C. A.; J. Neurosci.2009 , 29, 12079. Os resultados de estudos a respeito deste composto, porém, ainda são discrepantes, necessitando de estudos mais aprofundados.281281 Appleby, B. S.; Nacopoulos, D.; Milano, N.; Zhong, K.; Cummings, J. L.; Dementia Geriatr. Cognit. Disord.2013 , 35, 1.
CONSIDERAÇÕES FINAIS
Apesar da doença de Alzheimer representar a forma mais comum de demência em idosos, existindo cerca de 35 milhões de pessoas afetadas no mundo, ainda não há estudos concretos demonstrando a eficiência das drogas atualmente em uso. As hipóteses moleculares da doença de Alzheimer mais estudadas diferem a respeito de qual característica fisiopatológica é a mais importante neste contexto, levando a diferentes conclusões mecanísticas, e, consequentemente, a diferentes abordagens terapêuticas. É clara a existência de um "tempo de indução" entre o ostracismo "científico" de uma teoria e o seu abandono clínico. Por exemplo, apesar da falta de sucesso medicinal das drogas baseadas na hipótese colinérgica, três dos quatro fármacos atualmente disponíveis para o tratamento da DA fundamentam nela as suas ações terapêuticas. Neste cenário, de acordo com a constante evolução da compreensão da doença, e no atual fortalecimento das hipóteses metálica e do diabetes de tipo 3, compostos com esse tipo de ação aparentam ser os mais promissores, tendo como objetivo o tratamento das causas subjacentes e não somente dos sintomas da doença. Outra abordagem promissora é a de drogas híbridas, que têm a capacidade de se enquadrar em mais de uma hipótese etiológica.213213 Huang, W.; Wei, W.; Shen, Z.; RSC Adv.2014 , 4, 52088.
As pesquisas atuais abrangem cada vez mais a interdisciplinaridade, combinando estudos químicos, bioquímicos, biológicos e toxicológicos, permitindo uma compreensão mais completa dos mecanismos subjacentes e a formulação de terapias mais eficazes para compor o arsenal químico contra a doença de Alzheimer, que desafia a comunidade científica há mais de um século.
AGRADECIMENTOS
Os autores Rey, N. A. e De Falco, A. agradecem ao CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brasil) pelos auxílios e bolsas concedidos. Rey, N. A. é também grato à FAPERJ (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro) pela bolsa JCNE outorgada. Hauser-Davis, R. A. agradece à CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) pela bolsa concedida.
REFERÊNCIAS
-
1Burns, A.; Iliffe, S.; British Med. J.2009 , 338, b158.
-
2Reitz, C.; Brayne, C.; Mayeux, R.; Nat. Rev. Neurol.2011 , 7, 137.
-
3Ferri, C. P.; Revista Brasileira de Psiquiatria2012 , 34, 371.
-
4Herrera, E.; Caramelli, P.; Silveira, A. S.; Nitrini, R.;Alzheimer Dis. Assoc. Disord.2002 , 16, 103.
-
5Alzheimer, A.; Neurol. Central.1907 , 25, 1134.
-
6Small, G. W.; Rabins, P. V.; Barry, P. P.; Buckholtz, N. S.; DeKosky, S. T.; Ferris, S. H.; Finkel, S. I.; Gwyther, L. P.; Khachaturian, Z. S.; Lebowitz, B. D.; McRae, T. D.; Morris, J. C.; Oakley, F.; Schneider, L. S.; Streim, J. E.; Sunderland, T.; Teri, L. A.; Tune, L. E.; JAMA, J. Am. Med. Assoc.1997 , 278, 1363.
-
7Bekris, L. M.; Yu, C. E.; Bird, T. D.; Tsuang, D. W.;Journal of Geriatric Psychiatry and Neurology2010 , 23, 213.
-
8Serrano-Pozo, A.; Frosch, M. P.; Masliah, E.; Hyman, B. T.;Cold Spring Harbor Perspect. Biol.2011 , 1, a006189.
-
9Smith, M. A. C.; Revista Brasileira de Psiquiatria1999 , 21, 03.
-
10Hardman, J. G.; Limbird, L. E.; Gilman, A. G.; Goodman, L. S.; Gilman, A.; Goodman & Gilman's the pharmacological basis of therapeutics, McGraw-Hill: New York, 1996.
-
11Bartus, R. T.; Dean, R. L.; Beer, B.; Lippa, A. S.;Science1982 , 217, 408.
-
12Coyle, J. T.; Price, D. L.; DeLong, M. R.; Science1983 , 219, 1184.
-
13Deutsch, J. A.; Science1971 , 174, 788.
-
14Davies, P.; Maloney, A. J.; Lancet1976 , 2, 1403.
-
15Kása, P.; Rakonczay, Z.; Gulya, K.; Prog. Neurobiol.1997 , 52, 511.
-
16Wilcock, G. K.; Esiri, M. M.; Bowen, D. M.; Smith, C. C.; J. Neurol. Sci.1982 , 57, 407.
-
17Drachman, D. A.; Sahakian, B. J.; Arch. Neurol.1980 , 37, 674.
-
18Christie, J. E.; Shering, A.; Ferguson, J.; Glen, A. I.; Br. J. Psychiatry1981 , 138, 46.
-
19Bennett, B. M.; Reynolds, J. N.; Prusky, G. T.; Douglas, R. M.; Sutherland, R. J.; Thatcher, G. R.; Neuropsychopharmacology2007 , 32, 505.
-
20Muthuraju, S.; Maiti, P.; Solanki, P.; Sharma, A. K.; Amitabh; Singh, S. B.; Prasad, D.; Ilavazhagan, G.; Behav. Brain Res.2009 , 203, 1.
-
21Berger-Sweeney, J.; Arnold, A.; Gabeau, D.; Mills, J.;Behav. Neurosci.1995 , 109, 859.
-
22Hasselmo, M. E.; Curr. Opin. Neurobiol.2006 , 16, 710.
-
23Buccafusco, J. J.; Letchworth, S. R.; Bencherif, M.; Lippiello, P. M.; Trends Pharmacol. Sci.2005 , 26, 352.
-
24Green, A.; Ellis, K. A.; Ellis, J.; Bartholomeusz, C. F.; Ilic, S.; Croft, R. J.; Phan, K. L.; Nathan, P. J.; Pharmacol. Biochem. Behav.2005 , 81, 575.
-
25Greenamyre, J. T.; Maragos, W. F.; Albin, R. L.; Penney, J. B.; Young, A. B.; Prog. Neuro-Psychopharmacol. Biol. Psychiatry1988 , 12, 421.
-
26Danysz, W.; Parsons, C. G.; Mobius, H. J.; Stoffler, A.; Quack, G.;Neurotox. Res.2000 , 2, 85.
-
27Parsons, C. G.; Stöffler, A.; Danysz, W.;Neuropharmacology2007 , 53, 699.
-
28Sucher, N. J.; Awobuluyi, M.; Choi, Y. B.; Lipton, S. A.;Trends Pharmacol. Sci.1996 , 17, 348.
-
29Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S. F.;Pharmacol. Rev.1999 , 51, 7.
-
30Greenamyre, J. T.; Young, A. B.; Neurobiol. Aging1989 , 10, 593.
-
31Nie, Q.; Du, X. G.; Geng, M. Y.; Acta Pharmacol. Sin.2011 , 32, 545.
-
32Jenkins, E. C.; Devine-Gage, E. A.; Robakis, N. K.; Yao, X. L.; Brown, W. T.; Houck, G. E.; Wolfe, G.; Ramakrishna, N.; Silverman, W. P.; Wisniewski, H. M.; Biochem. Biophys. Res. Commun.1988 , 151, 1.
-
33Selkoe, D. J.; J. Biol. Chem.1996 , 271, 18295.
-
34Hardy, J. A.; Higgins, G. A.; Science1992 , 256, 184.
-
35Giaccone, G.; Tagliavini, F.; Linoli, G.; Bouras, C.; Frigerio, L.; Frangione, B.; Bugiani, O.; Neurosci. Lett.1989 , 97, 232.
-
36Iwatsubo, T.; Mann, D. M.; Odaka, A.; Suzuki, N.; Ihara, Y.;Ann. Neurol.1995 , 37, 294.
-
37Goate, A.; Chartier-Harlin, M. C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.;Nature1991 , 349, 704.
-
38Chartier-Harlin, M. C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.;Nature1991 , 353, 844.
-
39, J.; Farlow, M.; Ghetti, B.; Benson, M. D.;Science1991 , 254, 97.
-
40Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L.; Nat. Genet.1992 , 1, 345.
-
41Hendriks, L.; van Duijn, C. M.; Cras, P.; Cruts, M.; Van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M. G.; Antonarakis, S. E.; Martin, J. J.;Nat. Genet.1992 , 1, 218.
-
42Levy, E.; Carman, M. D.; Fernandez-Madrid, I. J.; Power, M. D.; Lieberburg, I.; van Duinen, S. G.; Bots, G. T.; Luyendijk, W.; Frangione, B.;Science1990 , 248, 1124.
-
43Haass, C.; Schlossmacher, M. G.; Hung, A. Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B. L.; Lieberburg, I.; Koo, E. H.; Schenk, D.; Teplow, D. B.; Nature1992 , 359, 322.
-
44Mattson, M. P.; Physiol. Rev.1997 , 77, 1081.
-
45Nitsch, R. M.; Farber, S. A.; Growdon, J. H.; Wurtman, R. J.;Proc. Natl. Acad. Sci. U. S. A.1993 , 90, 5191.
-
46Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A. Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D. J.;Nature1992 , 360, 672.
-
47Soreghan, B.; Kosmoski, J.; Glabe, C.; J. Biol. Chem.1994 , 269, 28551.
-
48Finckh, U.; Kuschel, C.; Anagnostouli, M.; Patsouris, E.; Pantes, G. V.; Gatzonis, S.; Kapaki, E.; Davaki, P.; Lamszus, K.; Stavrou, D.; Gal, A.;Neurogenetics2005 , 6, 85.
-
49Renbaum, P.; Levy-Lahad, E.; Cell. Mol. Life Sci.1998 , 54, 910.
-
50Tanzi, R. E.; Kovacs, D. M.; Kim, T. W.; Moir, R. D.; Guenette, S. Y.; Wasco, W.; Neurobiol Dis.1996 , 3, 159.
-
51Tanzi, R.; Gaston, S.; Bush, A.; Romano, D.; Pettingell, W.; Peppercorn, J.; Paradis, M.; Gurubhagavatula, S.; Jenkins, B.; Wasco, W.;Genetica1993 , 91, 255.
-
52Wasco, W.; Peppercorn, J.; Tanzi, R. E.; Ann. N. Y. Acad. Sci.1993 , 695, 203.
-
53Taylor, J. E.; Tinklenberg, J. R.; Eng, L. F.; Yesavage, J. A.; Vinogradov, S.; Davies, H. G.; Gonzalez-DeWhitt, P. A.; Frossard, P. M.;Mol. Biol. Med.1988 , 5, 167.
-
54Haass, C.; Hung, A. Y.; Selkoe, D. J.; Teplow, D. B.; J. Biol. Chem.1994 , 269, 17741.
-
55Spires, T. L.; Hyman, B. T.; NeuroRx2005 , 2, 423.
-
56Saido, T. C.; Iwatsubo, T.; Mann, D. M.; Shimada, H.; Ihara, Y.; Kawashima, S.; Neuron1995 , 14, 457.
-
57He, W.; Barrow, C. J.; Biochemistry1999 , 38, 10871.
-
58Tabaton, M.; Nunzi, M. G.; Xue, R.; Usiak, M.; Autiliogambetti, L.; Gambetti, P.; Biochem. Biophys. Res. Commun.1994 , 200, 1598.
-
59Gandy, S.; J. Clin. Invest.2005 , 115, 1121.
-
60Hane, F.; Leonenko, Z.; Biomolecules2014 , 4, 101.
-
61Gu, L.; Liu, C.; Guo, Z.; J. Biol. Chem.2013 , 288, 18673.
-
62Moores, B.; Drolle, E.; Attwood, S. J.; Simons, J.; Leonenko, Z.;PLoS One2011 , 6, e25954.
-
63Lee, G.; Lee, W.; Lee, H.; Woo Lee, S.; Sung Yoon, D.; Eom, K.; Kwon, T.; Appl. Phys. Lett.2012 , 101, 043703.
-
64Burke, K. A.; Yates, E. A.; Legleiter, J.; Front. Neurol.2013 , 4, 17.
-
65Hane, F.; Tran, G.; Attwood, S. J.; Leonenko, Z.; PLoS One2013 , 8, e59005.
-
66Lee, J.; Culyba, E. K.; Powers, E. T.; Kelly, J. W.; Nat. Chem. Biol.2011 , 7, 602.
-
67Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C. B.; Condron, M. M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D. B.; Younkin, S. G.; Näslund, J.; Lannfelt, L.; Nat. Neurosci.2001 , 4, 887.
-
68Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T. M.; Milton, S. C.; Hall, J. E.; Glabe, C. G.; J. Biol. Chem.2004 , 279, 46363.
-
69Klein, W. L.; Krafft, G. A.; Finch, C. E.; Trends Neurosci.2001 , 24, 219.
-
70Puzzo, D.; Vitolo, O.; Trinchese, F.; Jacob, J. P.; Palmeri, A.; Arancio, O.; J. Neurosci.2005 , 25, 6887.
-
71Selkoe, D. J.; Science2002 , 298, 789.
-
72Walsh, D. M.; Klyubin, I.; Fadeeva, J. V.; Cullen, W. K.; Anwyl, R.; Wolfe, M. S.; Rowan, M. J.; Selkoe, D. J.; Nature2002 , 416, 535.
-
73Wishik, C. M.; Novak, M.; Edwards, P. C.; Klug, A.; Tichelaar, W.; Crowther, R. A.; Proc. Natl. Acad. Sci. U. S. A.1988 , 85, 4884.
-
74Selkoe, D.; Mandelkow, E.; Holtzman, D.; Cold Spring Harbor Perspect. Med.2012 , 2, a011460.
-
75Zheng, W. H.; Bastianetto, S.; Mennicken, F.; Ma, W.; Kar, S.;Neuroscience2002 , 115, 201.
-
76Lloret, A.; Badia, M. C.; Giraldo, E.; Ermak, G.; Alonso, M. D.; Pallardó, F. V.; Davies, K. J.; Viña, J.; J. Alzheimer's Dis.2011 , 27, 701.
-
77Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B. A.;Neuron1995 , 14, 879.
-
78Stancu, I. C.; Vasconcelos, B.; Terwel, D.; Dewachter, I.;Mol. Neurodegener.2014 , 9, 51.
-
79Hu, X.; Li, X.; Zhao, M.; Gottesdiener, A.; Luo, W.; Paul, S.;Mol. Neurodegener.2014 , 9, 52.
-
80Annamalai, B.; Won, J. S.; Choi, S.; Singh, I.; Singh, A. K.;Biochem. Biophys. Res. Commun.2015 , 458, 214.
-
81Blennow, K.; de Leon, M. J.; Zetterberg, H.; Lancet2006 , 368, 387.
-
82Weiner, M. W.; Veitch, D. P.; Aisen, P. S.; Beckett, L. A.; Cairns, N. J.; Green, R. C.; Harvey, D.; Jack, C. R.; Jagust, W.; Liu, E.; Morris, J. C.; Petersen, R. C.; Saykin, A. J.; Schmidt, M. E.; Shaw, L.; Siuciak, J. A.; Soares, H.; Toga, A. W.; Trojanowski, J. Q.; Initiative, A. s. D. N.;Alzheimer's Dementia2012 , 8, S1.
-
83Mudher, A.; Lovestone, S.; Trends Neurosci.2002 , 25, 22.
-
84Urbanc, B.; Betnel, M.; Cruz, L.; Li, H.; Fradinger, E. A.; Monien, B. H.; Bitan, G.; J. Mol. Biol.2011 , 410, 316.
-
85Danysz, W.; Parsons, C. G.; Br. J. Pharmacol.2012 , 167, 324.
-
86Yamada, M.; Chiba, T.; Sasabe, J.; Nawa, M.; Tajima, H.; Niikura, T.; Terashita, K.; Aiso, S.; Kita, Y.; Matsuoka, M.; Nishimoto, I.;Behav. Brain Res.2005 , 164, 139.
-
87Beyreuther, K.; Multhaup, G.; Masters, C. L.; Ciba Found. Symp.1996 , 199, 119.
-
88Lambert, M. P.; Barlow, A. K.; Chromy, B. A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T. E.; Rozovsky, I.; Trommer, B.; Viola, K. L.; Wals, P.; Zhang, C.; Finch, C. E.; Krafft, G. A.; Klein, W. L.; Proc. Natl. Acad. Sci. U. S. A.1998 , 95, 6448.
-
89Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R. P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J. M.; Grüning, B. A.; Wang, Q.; Schmidt, M. R.; Lurz, R.; Anwyl, R.; Schnoegl, S.; Fändrich, M.; Frank, R. F.; Reif, B.; Günther, S.; Walsh, D. M.; Wanker, E. E.; Nat. Chem. Biol.2012 , 8, 93.
-
90Knobloch, M.; Farinelli, M.; Konietzko, U.; Nitsch, R. M.; Mansuy, I. M.; J. Neurosci.2007 , 27, 7648.
-
91Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O.; J. Neurosci.2008 , 28, 14537.
-
92Koffie, R. M.; Hyman, B. T.; Spires-Jones, T. L.; Mol. Neurodegener.2011 , 6, 63.
-
93Ferreira, S. T.; Klein, W. L.; Neurobiol. Learn. Mem.2011 , 96, 529.
-
94Selkoe, D. J.; Behav. Brain Res.2008 , 192, 106.
-
95Lue, L. F.; Kuo, Y. M.; Roher, A. E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J. H.; Rydel, R. E.; Rogers, J.; Am. J. Pathol.1999 , 155, 853.
-
96Lacor, P. N.; Buniel, M. C.; Chang, L.; Fernandez, S. J.; Gong, Y.; Viola, K. L.; Lambert, M. P.; Velasco, P. T.; Bigio, E. H.; Finch, C. E.; Krafft, G. A.; Klein, W. L.; J. Neurosci.2004 , 24, 10191.
-
97Selkoe, D. J.; Cold Spring Harbor Perspect. Biol.2011 , 3, a004457.
-
98Klein, W. L.; J. Alzheimer's Dis.2013 , 33, S49.
-
99Schnabel, J.; Nature2011 , 475, S12.
-
100Parikh, V.; Bernard, C. S.; Naughton, S. X.; Yegla, B.;Behav. Brain Res.2014 , 274, 30.
-
101Mohandas, E.; Rajmohan, V.; Raghunath, B.; Indian J. Psychiatry2009 , 51, 55.
-
102Kar, S.; Slowikowski, S. P.; Westaway, D.; Mount, H. T.; J. Psychiatry Neurosci.2004 , 29, 427.
-
103Yan, Z.; Feng, J.; Curr. Alzheimer Res.2004 , 1, 241.
-
104Auld, D. S.; Kornecook, T. J.; Bastianetto, S.; Quirion, R.;Prog. Neurobiol.2002 , 68, 209.
-
105Deibel, M. A.; Ehmann, W. D.; Markesbery, W. R.; J. Neurol. Sci.1996 , 143, 137.
-
106Azimi, S.; Rauk, A.; Int. J. Alzheimer's Dis.2011 , 2011, 539762.
-
107Tõugu, V.; Karafin, A.; Palumaa, P.; J. Neurochem.2008 , 104, 1249.
-
108Huang, X.; Atwood, C. S.; Moir, R. D.; Hartshorn, M. A.; Tanzi, R. E.; Bush, A. I.; J. Biol. Inorg. Chem.2004 , 9, 954.
-
109Craddock, T. J.; Tuszynski, J. A.; Chopra, D.; Casey, N.; Goldstein, L. E.; Hameroff, S. R.; Tanzi, R. E.; PLoS One2012 , 7, e33552.
-
110Miura, T.; Suzuki, K.; Kohata, N.; Takeuchi, H.;Biochemistry2000 , 39, 7024.
-
111Bush, A. I.; Pettingell, W. H.; Multhaup, G.; d Paradis, M.; Vonsattel, J. P.; Gusella, J. F.; Beyreuther, K.; Masters, C. L.; Tanzi, R. E.;Science1994 , 265, 1464.
-
112Huang, X.; Atwood, C. S.; Moir, R. D.; Hartshorn, M. A.; Vonsattel, J. P.; Tanzi, R. E.; Bush, A. I.; J. Biol. Chem.1997 , 272, 26464.
-
113Yang, D. S.; McLaurin, J.; Qin, K.; Westaway, D.; Fraser, P. E.;Eur. J. Biochem.2000 , 267, 6692.
-
114Smith, M. A.; Wehr, K.; Harris, P. L.; Siedlak, S. L.; Connor, J. R.; Perry, G.; Brain Res.1998 , 788, 232.
-
115Faller, P.; Hureau, C.; Chemistry2012 , 18, 15910.
-
116Danscher, G.; Jensen, K. B.; Frederickson, C. J.; Kemp, K.; Andreasen, A.; Juhl, S.; Stoltenberg, M.; Ravid, R.; J. Neurosci. Methods1997 , 76, 53.
-
117Lovell, M. A.; Robertson, J. D.; Teesdale, W. J.; Campbell, J. L.; Markesbery, W. R.; J. Neurol. Sci.1998 , 158, 47.
-
118Sayre, L. M.; Perry, G.; Harris, P. L.; Liu, Y.; Schubert, K. A.; Smith, M. A.; J. Neurochem.2000 , 74, 270.
-
119Zhu, X.; Su, B.; Wang, X.; Smith, M. A.; Perry, G.; Cell. Mol. Life Sci.2007 , 64, 2202.
-
120Bonda, D. J.; Lee, H. G.; Blair, J. A.; Zhu, X.; Perry, G.; Smith, M. A.; Metallomics2011 , 3, 267.
-
121Barreiros, A. L. B. S.; David, J. M.; David, J. P.; Quim. Nova2006 , 29, 113.
-
122Barnham, K. J.; Bush, A. I.; Curr. Opin. Chem. Biol.2008 , 12, 222.
-
123Barnham, K. J.; Masters, C. L.; Bush, A. I.; Nat. Rev. Drug Discov.2004 , 3, 205.
-
124Su, B.; Wang, X.; Nunomura, A.; Moreira, P. I.; Lee, H.; Perry, G.; Smith, M. A.; Zhu, X.; Curr. Alzheimer Res.2008 , 5, 525.
-
125Liu, G.; Men, P.; Kudo, W.; Perry, G.; Smith, M. A.;Neurosci. Lett.2009 , 455, 187.
-
126Adlard, P. A.; Parncutt, J. M.; Finkelstein, D. I.; Bush, A. I.;J. Neurosci.2010 , 30, 1631.
-
127Lee, J. Y.; Cole, T. B.; Palmiter, R. D.; Suh, S. W.; Koh, J. Y.;Proc. Natl. Acad. Sci. U. S. A.2002 , 99, 7705.
-
128Cole, T. B.; Wenzel, H. J.; Kafer, K. E.; Schwartzkroin, P. A.; Palmiter, R. D.; Proc. Natl. Acad. Sci. U. S. A.1999 , 96, 1716.
-
129Bush, A. I.; J. Alzheimer's Dis.2013 , 33, S277.
-
130Miller, L. M.; Wang, Q.; Telivala, T. P.; Smith, R. J.; Lanzirotti, A.; Miklossy, J.; J. Struct. Biol.2006 , 155, 30.
-
131Miller, Y.; Ma, B.; Nussinov, R.; Proc. Natl. Acad. Sci. U. S. A.2010 , 107, 9490.
-
132Hoernke, M.; Koksch, B.; Brezesinski, G.; Biophys. Chem.2010 , 150, 64.
-
133Marino, T.; Russo, N.; Toscano, M.; Pavelka, M.;Interdiscip. Sci.2010 , 2, 57.
-
134Leal, M. F. C.; Catarino, R. I. L.; Pimenta, A. M.; Souto, M. R. S.; Pinheiro, T. S. N.; Quim. Nova2012 , 35, 1985.
-
135Adlard, P. A.; Bica, L.; White, A. R.; Nurjono, M.; Filiz, G.; Crouch, P. J.; Donnelly, P. S.; Cappai, R.; Finkelstein, D. I.; Bush, A. I.;PLoS One2011 , 6, e17669.
-
136Friedlich, A. L.; Lee, J. Y.; van Groen, T.; Cherny, R. A.; Volitakis, I.; Cole, T. B.; Palmiter, R. D.; Koh, J. Y.; Bush, A. I.; J. Neurosci.2004 , 24, 3453.
-
137Sensi, S. L.; Paoletti, P.; Koh, J. Y.; Aizenman, E.; Bush, A. I.; Hershfinkel, M.; J. Neurosci.2011 , 31, 16076.
-
138Smith, M. A.; Harris, P. L.; Sayre, L. M.; Perry, G.; Proc. Natl. Acad. Sci. U. S. A.1997 , 94, 9866.
-
139Lei, P.; Ayton, S.; Finkelstein, D. I.; Spoerri, L.; Ciccotosto, G. D.; Wright, D. K.; Wong, B. X.; Adlard, P. A.; Cherny, R. A.; Lam, L. Q.; Roberts, B. R.; Volitakis, I.; Egan, G. F.; McLean, C. A.; Cappai, R.; Duce, J. A.; Bush, A. I.; Nat. Med.2012 , 18, 291.
-
140Shcherbatykh, I.; Carpenter, D. O.; J. Alzheimer's Dis.2007 , 11, 91.
-
141Frisardi, V.; Solfrizzi, V.; Capurso, C.; Kehoe, P. G.; Imbimbo, B. P.; Santamato, A.; Dellegrazie, F.; Seripa, D.; Pilotto, A.; Capurso, A.; Panza, F.; J. Alzheimer's Dis.2010 , 20, 17.
-
142Bakulski, K. M.; Rozek, L. S.; Dolinoy, D. C.; Paulson, H. L.; Hu, H.; Curr. Alzheimer Res.2012 , 9, 563.
-
143von Lindern, I.; Spalinger, S.; Petroysan, V.; von Braun, M.;Sci. Total Environ.2003 , 303, 139.
-
144Lewin, M. D.; Sarasua, S.; Jones, P. A.; Environ. Res.1999 , 81, 52.
-
145Brown, D. R.; Dalton Trans.2009 , 4069.
-
146Levi, R.; Wolf, T.; Fleminger, G.; Solomon, B.; Mol. Cell. Biochem.1998 , 189, 41.
-
147Kawahara, M.; Kato-Negishi, M.; Int. J. Alzheimer's Dis.2011 , 2011, 1.
-
148Zatta, P.; Ibn-Lkhayat-Idrissi, M.; Zambenedetti, P.; Kilyen, M.; Kiss, T.; Brain Res. Bull.2002 , 59, 41.
-
149Savory, J.; Herman, M. M.; Ghribi, O.; J. Alzheimer's Dis.2006 , 10, 135.
-
150Basha, M. R.; Wei, W.; Bakheet, S. A.; Benitez, N.; Siddiqi, H. K.; Ge, Y. W.; Lahiri, D. K.; Zawia, N. H.; J. Neurosci.2005 , 25, 823.
-
151Wu, J.; Basha, M. R.; Brock, B.; Cox, D. P.; Cardozo-Pelaez, F.; McPherson, C. A.; Harry, J.; Rice, D. C.; Maloney, B.; Chen, D.; Lahiri, D. K.; Zawia, N. H.; J. Neurosci.2008 , 28, 3.
-
152Bolin, C. M.; Basha, R.; Cox, D.; Zawia, N. H.; Maloney, B.; Lahiri, D. K.; Cardozo-Pelaez, F.; FASEB J.2006 , 20, 788.
-
153Pendergrass, A. C.; Haley, B. E.; Vimy, M. J.; Winfield, S. A.; Lorscheider, F. L.; Neurotoxicology1997 , 18, 315.
-
154Leong, C. C. W.; Syed, N. I.; Lorscheider, F. L.;Neuroreport2001 , 12, 733.
-
155Haley, B. E.; Medical Veritas2007 , 4, 1510.
-
156Santos, J. R.; Gois, A. M.; Mendonça, D. M.; Freire, M. A.;Front Aging Neurosci.2014 , 6, 206.
-
157Cardoso, B. R.; Roberts, B. R.; Bush, A. I.; Hare, D. J.;Metallomics2015 , 7, 1213.
-
158González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J. L.;Biometals2014 , 27, 539.
-
159Rita Cardoso, B.; Silva Bandeira, V.; Jacob-Filho, W.; Franciscato Cozzolino, S. M.; J. Trace Elem. Med. Biol.2014 , 28, 422.
-
160Olde Rikkert, M. G.; Verhey, F. R.; Sijben, J. W.; Bouwman, F. H.; Dautzenberg, P. L.; Lansink, M.; Sipers, W. M.; van Asselt, D. Z.; van Hees, A. M.; Stevens, M.; Vellas, B.; Scheltens, P.; J. Alzheimer's Dis.2014 , 41, 261.
-
161Haratake, M.; Yoshida, S.; Mandai, M.; Fuchigami, T.; Nakayama, M.;Metallomics2013 , 5, 479.
-
162van Eersel, J.; Ke, Y. D.; Liu, X.; Delerue, F.; Kril, J. J.; Götz, J.; Ittner, L. M.; Proc. Natl. Acad. Sci. U. S. A.2010 , 107, 13888.
-
163Corcoran, N. M.; Martin, D.; Hutter-Paier, B.; Windisch, M.; Nguyen, T.; Nheu, L.; Sundstrom, L. E.; Costello, A. J.; Hovens, C. M.; J. Clin. Neurosci.2010 , 17, 1025.
-
164Wang, Z.; Wang, Y.; Li, W.; Mao, F.; Sun, Y.; Huang, L.; Li, X.;ACS Chem. Neurosci.2014 , 5, 952.
-
165Ferreira, S. T.; Clarke, J. R.; Bomfim, T. R.; De Felice, F. G.;Alzheimer's Dementia2014 , 10, S76.
-
166Lourenco, M. V.; Ferreira, S. T.; De Felice, F. G.; Prog. Neurobiol.2015 , 129, 37.
-
167Ott, A.; Stolk, R. P.; van Harskamp, F.; Pols, H. A.; Hofman, A.; Breteler, M. M.; Neurology1999 , 53, 1937.
-
168Beydoun, M. A.; Lhotsky, A.; Wang, Y.; Dal Forno, G.; An, Y.; Metter, E. J.; Ferrucci, L.; O'Brien, R.; Zonderman, A. B.; Am. J. Epidemiol.2008 , 168, 1179.
-
169Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L.;Neurology2008 , 71, 1065.
-
170Pfrieger, F. W.; Cell. Mol. Life Sci.2003 , 60, 1158.
-
171Wernette-Hammond, M. E.; Lauer, S. J.; Corsini, A.; Walker, D.; Taylor, J. M.; Rall, S. C.; J. Biol. Chem.1989 , 264, 9094.
-
172Rebeck, G. W.; Reiter, J. S.; Strickland, D. K.; Hyman, B. T.;Neuron1993 , 11, 575.
-
173Schmechel, D. E.; Saunders, A. M.; Strittmatter, W. J.; Crain, B. J.; Hulette, C. M.; Joo, S. H.; Pericak-Vance, M. A.; Goldgaber, D.; Roses, A. D.; Proc. Natl. Acad. Sci. U. S. A.1993 , 90, 9649.
-
174Tiraboschi, P.; Hansen, L. A.; Masliah, E.; Alford, M.; Thal, L. J.; Corey-Bloom, J.; Neurology2004 , 62, 1977.
-
175Benjamin, R.; Leake, A.; Ince, P. G.; Perry, R. H.; McKeith, I. G.; Edwardson, J. A.; Morris, C. M.; Neurodegen.1995 , 4, 443.
-
176Heinonen, O.; Lehtovirta, M.; Soininen, H.; Helisalmi, S.; Mannermaa, A.; Sorvari, H.; Kosunen, O.; Paljärvi, L.; Ryynänen, M.; Riekkinen, P. J.; J. Neurobiol. Aging1995 , 16, 505.
-
177Itoh, Y.; Yamada, M.; N. Engl. J. Med.1996 , 334, 599.
-
178Landén, M.; Thorsell, A.; Wallin, A.; Blennow, K.; J. Neurol., Neurosurg. Psychiatry1996 , 61, 352.
-
179Wisniewski, T.; Frangione, B.; Neurosci. Lett.1992 , 135, 235.
-
180Reiman, E. M.; Chen, K.; Liu, X.; Bandy, D.; Yu, M.; Lee, W.; Ayutyanont, N.; Keppler, J.; Reeder, S. A.; Langbaum, J. B.; Alexander, G. E.; Klunk, W. E.; Mathis, C. A.; Price, J. C.; Aizenstein, H. J.; DeKosky, S. T.; Caselli, R. J.; Proc. Natl. Acad. Sci. U. S. A.2009 , 106, 6820.
-
181Sunderland, T.; Mirza, N.; Putnam, K. T.; Linker, G.; Bhupali, D.; Durham, R.; Soares, H.; Kimmel, L.; Friedman, D.; Bergeson, J.; Csako, G.; Levy, J. A.; Bartko, J. J.; Cohen, R. M.; Biol. Psychiatry2004 , 56, 670.
-
182Steen, E.; Terry, B. M.; Rivera, E. J.; Cannon, J. L.; Neely, T. R.; Tavares, R.; Xu, X. J.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2005 , 7, 63.
-
183Rivera, E. J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2005 , 8, 247.
-
184Braak, H.; Braak, E.; Acta Neuropathol.1991 , 82, 239.
-
185de la Monte, S. M.; Tong, M.; Lester-Coll, N.; Plater, M.; Wands, J. R.; J. Alzheimer's Dis.2006 , 10, 89.
-
186De Felice, F. G.; Lourenco, M. V.; Ferreira, S. T.;Alzheimer's Dementia2014 , 10, S26.
-
187Lester-Coll, N.; Rivera, E. J.; Soscia, S. J.; Doiron, K.; Wands, J. R.; de la Monte, S. M.; J. Alzheimer's Dis.2006 , 9, 13.
-
188Craft, S.; Watson, G. S.; Lancet Neurol.2004 , 3, 169.
-
189Reger, M. A.; Watson, G. S.; Green, P. S.; Wilkinson, C. W.; Baker, L. D.; Cholerton, B.; Fishel, M. A.; Plymate, S. R.; Breitner, J. C.; DeGroodt, W.; Mehta, P.; Craft, S.; Neurology2008 , 70, 440.
-
190Park, C. R.; Seeley, R. J.; Craft, S.; Woods, S. C.;Physiol. Behav.2000 , 68, 509.
-
191Bomfim, T. R.; Forny-Germano, L.; Sathler, L. B.; Brito-Moreira, J.; Houzel, J. C.; Decker, H.; Silverman, M. A.; Kazi, H.; Melo, H. M.; McClean, P. L.; Holscher, C.; Arnold, S. E.; Talbot, K.; Klein, W. L.; Munoz, D. P.; Ferreira, S. T.; De Felice, F. G.; J. Clin. Invest.2012 , 122, 1339.
-
192Talbot, K.; Wang, H. Y.; Kazi, H.; Han, L. Y.; Bakshi, K. P.; Stucky, A.; Fuino, R. L.; Kawaguchi, K. R.; Samoyedny, A. J.; Wilson, R. S.; Arvanitakis, Z.; Schneider, J. A.; Wolf, B. A.; Bennett, D. A.; Trojanowski, J. Q.; Arnold, S. E.; J. Clin. Invest.2012 , 122, 1316.
-
193de la Monte, S. M.; Drugs2012 , 72, 49.
-
194Anand, P.; Singh, B.; Arch. Pharm. Res.2013 , 36, 375.
-
195Cunningham, E. L.; Passmore, A. P.; Maturitas2013 , 76, 260.
-
196Ellul, J.; Archer, N.; Foy, C. M.; Poppe, M.; Boothby, H.; Nicholas, H.; Brown, R. G.; Lovestone, S.; J. Neurol., Neurosurg. Psychiatry2007 , 78, 233.
-
197Rodrigues Simões, M. C.; Dias Viegas, F. P.; Moreira, M. S.; de Freitas Silva, M.; Riquiel, M. M.; da Rosa, P. M.; Castelli, M. R.; dos Santos, M. H.; Soares, M. G.; Viegas, C.; Mini Rev. Med. Chem.2014 , 14, 2.
-
198Barnes, D. E.; Yaffe, K.; N. Engl. J. Med.2005 , 353, 951.
-
199Michaelson, D. M.; Alzheimer's Dementia2014 , 10, 861.
-
200Molino, I.; Colucci, L.; Fasanaro, A. M.; Traini, E.; Amenta, F.;Sci. World J.2013 , 2013, 925702.
-
201Trevisan, M. T. S.; Macedo, F. V. V.; Meent, M. v. d.; Rhee, I. K.; Verpoorte, R.; Quim. Nova2003 , 26, 301.
-
202Viegas Junior, C.; Bolzani, V. d. S.; Furlan, M.; Fraga, C. A. M.; Barreiro, E. J.; Quim. Nova2004 , 27, 655.
-
203Finefrock, A. E.; Bush, A. I.; Doraiswamy, P. M.; J. Am. Geriatr. Soc.2003 , 51, 1143.
-
204Bush, A. I.; Trends Neurosci.2003 , 26, 207.
-
205Pardridge, W. M.; Alzheimer's Dementia2009 , 5, 427.
-
206Dedeoglu, A.; Cormier, K.; Payton, S.; Tseitlin, K. A.; Kremsky, J. N.; Lai, L.; Li, X.; Moir, R. D.; Tanzi, R. E.; Bush, A. I.; Kowall, N. W.; Rogers, J. T.; Huang, X.; Exp. Gerontol.2004 , 39, 1641.
-
207Keberle, H.; Ann. N. Y. Acad. Sci.1964 , 119, 758.
-
208Cuajungco, M. P.; Fagét, K. Y.; Huang, X.; Tanzi, R. E.; Bush, A. I.; Ann. N. Y. Acad. Sci.2000 , 920, 292.
-
209Richardson, D. R.; Ponka, P.; Am. J. Hematol.1998 , 58, 299.
-
210Olivieri, N. F.; Brittenham, G. M.; Blood1997 , 89, 739.
-
211Kontoghiorghes, G. J.; Analyst1995 , 120, 845.
-
212Reichard, P.; Annu. Rev. Biochem.1988 , 57, 349.
-
213Huang, W.; Wei, W.; Shen, Z.; RSC Adv.2014 , 4, 52088.
-
214Kupershmidt, L.; Amit, T.; Bar-Am, O.; Weinreb, O.; Youdim, M. B.;Mol. Neurobiol.2012 , 46, 217.
-
215Zheng, H.; Gal, S.; Weiner, L. M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M. B.; J. Neurochem.2005 , 95, 68.
-
216Lee, S.; Zheng, X.; Krishnamoorthy, J.; Savelieff, M. G.; Park, H. M.; Brender, J. R.; Kim, J. H.; Derrick, J. S.; Kochi, A.; Lee, H. J.; Kim, C.; Ramamoorthy, A.; Bowers, M. T.; Lim, M. H.; J. Am. Chem. Soc.2014 , 136, 299.
-
217Sampson, E. L.; Jenagaratnam, L.; McShane, R.; Cochrane Database of Systematic Reviews2012 , 5, CD005380.
-
218Cherny, R. A.; Atwood, C. S.; Xilinas, M. E.; Gray, D. N.; Jones, W. D.; McLean, C. A.; Barnham, K. J.; Volitakis, I.; Fraser, F. W.; Kim, Y.; Huang, X.; Goldstein, L. E.; Moir, R. D.; Lim, J. T.; Beyreuther, K.; Zheng, H.; Tanzi, R. E.; Masters, C. L.; Bush, A. I.; Neuron2001 , 30, 665.
-
219Melov, S.; Trend Neurosci.2002 , 25, 121.
-
220Doraiswamy, P. M.; Finefrock, A. E.; Lancet Neurol.2004 , 3, 431.
-
221Bareggi, S. R.; Cornelli, U.; CNS Neurosci. Ther.2012 , 18, 41.
-
222Regland, B.; Lehmann, W.; Abedini, I.; Blennow, K.; Jonsson, M.; Karlsson, I.; Sjögren, M.; Wallin, A.; Xilinas, M.; Gottfries, C. G.;Dementia Geriatr. Cognit. Disord.2001 , 12, 408.
-
223Budimir, A.; Acta Pharm.2011 , 61, 1.
-
224Mancino, A. M.; Hindo, S. S.; Kochi, A.; Lim, M. H.; Inorg. Chem.2009 , 48, 9596.
-
225Levine, H.; Dingb, Q.; Walkerc, J. A.; Vossc, R. A.; Augelli-Szafrand, C. E.; Neurosci. Lett.2009 , 465, 99.
-
226Adlard, P. A.; Cherny, R. A.; Finkelstein, D. I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J. P.; Perez, K.; Laughton, K.; Li, Q. X.; Charman, S. A.; Nicolazzo, J. A.; Wilkins, S.; Deleva, K.; Lynch, T.; Kok, G.; Ritchie, C. W.; Tanzi, R. E.; Cappai, R.; Masters, C. L.; Barnham, K. J.; Bush, A. I.; Neuron2008 , 59, 43.
-
227Bush, A. I.; J. Alzheimer's Dis.2008 , 15, 223.
-
228Faux, N. G.; Ritchie, C. W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; Ingelsson, M.; Masters, C. L.; Tanzi, R. E.; Cummings, J. L.; Herd, C. M.; Bush, A. I.;J. Alzheimer's Dis.2010 , 20, 509.
-
229Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C. L.; Targum, S.; Bush, A. I.; Murdoch, R.; Wilson, J.; Ritchie, C. W.; Lancet Neurol.2008 , 7, 779.
-
230Crouch, P. J.; Savva, M. S.; Hung, L. W.; Donnelly, P. S.; Mot, A. I.; Parker, S. J.; Greenough, M. A.; Volitakis, I.; Adlard, P. A.; Cherny, R. A.; Masters, C. L.; Bush, A. I.; Barnham, K. J.; White, A. R.; J. Neurochem.2011 , 119, 220.
-
231Crouch, P. J.; Barnham, K. J.; Acc. Chem. Res.2012 , 45, 1604.
-
232Gomes, L. M.; Vieira, R. P.; Jones, M. R.; Wang, M. C.; Dyrager, C.; Souza-Fagundes, E. M.; Da Silva, J. G.; Storr, T.; Beraldo, H.; J. Inorg. Biochem.2014 , 139, 106.
-
233de Freitas, L. V.; da Silva, C. C.; Ellena, J.; Costa, L. A.; Rey, N. A.; Spectrochim. Acta, Part A2013 , 116, 41.
-
234Hauser-Davis, R. A.; de Freitas, L. V.; Cukierman, D. S.; Cruz, W. S.; Miotto, M. C.; Landeira-Fernandez, J.; Valiente-Gabioud, A. A.; Fernández, C. O.; Rey, N. A.; Metallomics2015 , 7, 743.
-
235Akter, K.; Lanza, E. A.; Martin, S. A.; Myronyuk, N.; Rua, M.; Raffa, R. B.; Br. J. Clin. Pharmacol.2011 , 71, 365.
-
236Lyketsos, C. G.; Steinberg, M.; Tschanz, J. T.; Norton, M. C.; Steffens, D. C.; Breitner, J. C.; Am. J. Psychiatry2000 , 157, 708.
-
237Kessing, L. V.; Harhoff, M.; Andersen, P. K.; International Psychogeriatrics2007 , 19, 902.
-
238Reifler, B. V.; Teri, L.; Raskind, M.; Veith, R.; Barnes, R.; White, E.; McLean, P.; Am. J. Psychiatry1989 , 146, 45.
-
239Nyth, A. L.; Gottfries, C. G.; Br. J. Psychiatry1990 , 157, 894.
-
240Magai, C.; Kennedy, G.; Cohen, C. I.; Gomberg, D.; American Journal of Geriatric Psychiatry2000 , 8, 66.
-
241Caltagirone, C.; Bianchetti, A.; Di Luca, M.; Mecocci, P.; Padovani, A.; Pirfo, E.; Scapicchio, P.; Senin, U.; Trabucchi, M.; Musicco, M.;Drugs Aging2005 , 22, 1.
-
242Lyketsos, C. G.; DelCampo, L.; Steinberg, M.; Miles, Q.; Steele, C. D.; Munro, C.; Baker, A. S.; Sheppard, J. M.; Frangakis, C.; Brandt, J.; Rabins, P. V.; Arch. Gen. Psychiatry2003 , 60, 737.
-
243Petracca, G. M.; Chemerinski, E.; Starkstein, S. E.;International Psychogeriatrics2001 , 13, 233.
-
244Nyth, A. L.; Gottfries, C. G.; Lyby, K.; Smedegaard-Andersen, L.; Gylding-Sabroe, J.; Kristensen, M.; Refsum, H. E.; Ofsti, E.; Eriksson, S.; Syversen, S.; Acta Psychiatr. Scand.1992 , 86, 138.
-
245Munro, C. A.; Brandt, J.; Sheppard, J. M.; Steele, C. D.; Samus, Q. M.; Steinberg, M.; Rabins, P. V.; Lyketsos, C. G.; American Journal of Geriatric Psychiatry2004 , 12, 491.
-
246Sheline, Y. I.; West, T.; Yarasheski, K.; Swarm, R.; Jasielec, M. S.; Fisher, J. R.; Ficker, W. D.; Yan, P.; Xiong, C.; Frederiksen, C.; Grzelak, M. V.; Chott, R.; Bateman, R. J.; Morris, J. C.; Mintun, M. A.; Lee, J. M.; Cirrito, J. R.; Sci. Transl. Med.2014 , 6, 236re4.
-
247Adelman, A.; J. Fam. Pract.1997 , 45, 98.
-
248Spencer, A. P.; Carson, D. S.; Crouch, M. A.; Arch. Intern. Med.1999 , 159, 1313.
-
249Dysken, M. W.; Sano, M.; Asthana, S.; Vertrees, J. E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G. D.; McCarten, J. R.; Malphurs, J.; Prieto, S.; Chen, P.; Loreck, D. J.; Trapp, G.; Bakshi, R. S.; Mintzer, J. E.; Heidebrink, J. L.; Vidal-Cardona, A.; Arroyo, L. M.; Cruz, A. R.; Zachariah, S.; Kowall, N. W.; Chopra, M. P.; Craft, S.; Thielke, S.; Turvey, C. L.; Woodman, C.; Monnell, K. A.; Gordon, K.; Tomaska, J.; Segal, Y.; Peduzzi, P. N.; Guarino, P. D.; JAMA2014 , 311, 33.
-
250Beal, M. F.; J. Bioenerg. Biomembr.2004 , 36, 381.
-
251Yang, X.; Yang, Y.; Li, G.; Wang, J.; Yang, E. S.; J. Mol. Neurosci.2008 , 34, 165.
-
252Wadsworth, T. L.; Bishop, J. A.; Pappu, A. S.; Woltjer, R. L.; Quinn, J. F.; J. Alzheimer's Dis.2008 , 14, 225.
-
253Ma, D.; Stokes, K.; Mahngar, K.; Domazet-Damjanov, D.; Sikorska, M.; Pandey, S.; Mitochondrion2014 , 17, 106.
-
254Galasko, D. R.; Peskind, E.; Clark, C. M.; Quinn, J. F.; Ringman, J. M.; Jicha, G. A.; Cotman, C.; Cottrell, B.; Montine, T. J.; Thomas, R. G.; Aisen, P.; Study, A. s. D. C.; Arch. Neurol.2012 , 69, 836.
-
255Smith, A. R.; Shenvi, S. V.; Widlansky, M.; Suh, J. H.; Hagen, T. M.; Curr. Med. Chem.2004 , 11, 1135.
-
256Cotman, C. W.; Head, E.; J. Alzheimer's Dis.2008 , 15, 685.
-
257Quinn, J. F.; Bussiere, J. R.; Hammond, R. S.; Montine, T. J.; Henson, E.; Jones, R. E.; Stackman, R. W.; Neurobiol. Aging2007 , 28, 213.
-
258Siedlak, S. L.; Casadesus, G.; Webber, K. M.; Pappolla, M. A.; Atwood, C. S.; Smith, M. A.; Perry, G.; Free Radic. Res.2009 , 43, 156.
-
259Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G.;J. Neural Transm., Suppl.2007 , 189.
-
260Smid, S. D.; Maag, J. L.; Musgrave, I. F.; Food Funct.2012 , 3, 1242.
-
261Richard, T.; Pawlus, A. D.; Iglésias, M. L.; Pedrot, E.; Waffo-Teguo, P.; Mérillon, J. M.; Monti, J. P.; Ann. N. Y. Acad. Sci.2011 , 1215, 103.
-
262Regitz, C.; Fitzenberger, E.; Mahn, F. L.; Dußling, L. M.; Wenzel, U.; Eur. J. Nutr.2015 ,
-
263Rege, S. D.; Geetha, T.; Broderick, T. L.; Babu, J. R.;Curr. Alzheimer Res.2015 , 12, 147.
-
264Bertelli, A. A.; Ferrara, F.; Diana, G.; Fulgenzi, A.; Corsi, M.; Ponti, W.; Ferrero, M. E.; Bertelli, A.; Int. J. Tissue React.1999 , 21, 93.
-
265Mercer, L. D.; Kelly, B. L.; Horne, M. K.; Beart, P. M.;Biochem. Pharmacol.2005 , 69, 339.
-
266Selvaraj, K.; Chowdhury, R.; Bhattacharjee, C.; Int. J. Pharm. Pharm. Sci.2013 , 5,
-
267Heo, H. J.; Lee, C. Y.; J. Agric. Food Chem.2004 , 52, 7514.
-
268Ansari, M. A.; Abdul, H. M.; Joshi, G.; Opii, W. O.; Butterfield, D. A.; J. Nutr. Biochem.2009 , 20, 269.
-
269Dobrowolska, J. A.; Michener, M. S.; Wu, G.; Patterson, B. W.; Chott, R.; Ovod, V.; Pyatkivskyy, Y.; Wildsmith, K. R.; Kasten, T.; Mathers, P.; Dancho, M.; Lennox, C.; Smith, B. E.; Gilberto, D.; McLoughlin, D.; Holder, D. J.; Stamford, A. W.; Yarasheski, K. E.; Kennedy, M. E.; Savage, M. J.; Bateman, R. J.; J. Neurosci.2014 , 34, 8336.
-
270Jacobsen, H.; Ozmen, L.; Caruso, A.; Narquizian, R.; Hilpert, H.; Jacobsen, B.; Terwel, D.; Tanghe, A.; Bohrmann, B.; J. Neurosci.2014 , 34, 11621.
-
271Patey, S. J.; Edwards, E. A.; Yates, E. A.; Turnbull, J. E.;J. Med. Chem.2006 , 49, 6129.
-
272Scholefield, Z.; Yates, E. A.; Wayne, G.; Amour, A.; McDowell, W.; Turnbull, J. E.; J. Cell. Biol.2003 , 163, 97.
-
273Menting, K. W.; Claassen, J. A.; Front. Aging Neurosci.2014 , 6, 165.
-
274Imbimbo, B. P.; Giardina, G. A.; Curr. Top. Med. Chem.2011 , 11, 1555.
-
275Plucińska, K.; Crouch, B.; Koss, D.; Robinson, L.; Siebrecht, M.; Riedel, G.; Platt, B.; J. Neurosci.2014 , 34, 10710.
-
276Wischik, C. M.; Edwards, P. C.; Lai, R. Y.; Roth, M.; Harrington, C. R.; Proc. Natl. Acad. Sci. U. S. A.1996 , 93, 11213.
-
277Callaway, N. L.; Riha, P. D.; Bruchey, A. K.; Munshi, Z.; Gonzalez-Lima, F.; Pharmacol. Biochem. Behav.2004 , 77, 175.
-
278Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M.; J. Biol. Chem.2005 , 280, 7614.
-
279Congdon, E. E.; Wu, J. W.; Myeku, N.; Figueroa, Y. H.; Herman, M.; Marinec, P. S.; Gestwicki, J. E.; Dickey, C. A.; Yu, W. H.; Duff, K. E.;Autophagy2012 , 8, 609.
-
280Jinwal, U. K.; Miyata, Y.; Koren, J.; Jones, J. R.; Trotter, J. H.; Chang, L.; O'Leary, J.; Morgan, D.; Lee, D. C.; Shults, C. L.; Rousaki, A.; Weeber, E. J.; Zuiderweg, E. R.; Gestwicki, J. E.; Dickey, C. A.; J. Neurosci.2009 , 29, 12079.
-
281Appleby, B. S.; Nacopoulos, D.; Milano, N.; Zhong, K.; Cummings, J. L.; Dementia Geriatr. Cognit. Disord.2013 , 35, 1.
Datas de Publicação
-
Publicação nesta coleção
Jan 2016
Histórico
-
Recebido
22 Jun 2015 -
Aceito
04 Ago 2015