Resumo
This work presents two amperometric methods for Sildenafil Citrate (SC) determination in erectile dysfunction drugs (Viagra® and Generics), using Batch Injection Analysis (BIA) and Flow Injection Analysis (FIA) systems with a cell for Screen Printed Electrodes (SPE). In Britton- Robinson buffer solution (pH 2), the SC presented an irreversible anodic peak at around +1.0 V vs. Ag pseudo-reference electrode, using five different working electrodes of commercial SPE. The carbon SPE (unmodified) was selected as the best working electrode, providing good sensitivity, precision and low-cost. The SC amperometric detection for two proposed methods was performed at +1.1 V. The best conditions for SC determination by hydrodynamic systems were obtained through injection volumes of 50 and 150 µL at flow rates of 100 and 75 µL s-1, for BIA and FIA respectively. In these conditions, BIA system showed the best sensitivity (0.188 µA mol-1 L) and theoretical analytical frequency (360 h-1) for SC determination. The repeatability studies were similar for both systems with relative standard deviation of approximately 3.0%. The SC analyses in pharmaceutical samples obtained by two proposed methods were compared to HPLC-UV method.
Keywords:
Sildenafil Citrate; Viagra®; FIA; BIA; Screen-Printed Electrode
Keywords:
Sildenafil Citrate; Viagra®; FIA; BIA; Screen-Printed Electrode
INTRODUÇÃO
O citrato de sildenafila (CS) é o princípio ativo do Viagra® (Pfizer), cujo medicamento é o mais usado no mundo para o tratamento da disfunção erétil masculina. No organismo, o CS inibe a enzima fosfodiesterase tipo 5 (PHD5), causando o aumento na circulação sanguínea e o relaxamento muscular peniano.11 Terrett, N. K.; Bell, A. S.; Brown, D.; Ellis, P.; Bioorg. Med. Chem. Lett. 1996, 6, 1819. Após expirar a patente do Viagra® no Brasil em 2010, vários laboratórios passaram a produzir medicamentos genéricos e similares com CS, além de suas formulações manipuladas pelas farmácias magistrais. Apesar da redução do custo dos medicamentos em virtude desta concorrência, a aquisição de formulações falsificadas e contrabandeadas no mercado negro é um problema recorrente no Brasil, com predominância para o medicamento Pramil fabricado no Paraguai.22 Hurtado, R. L.; Lasmar, M. C.; Cad. Saúde Pública 2014, 30, 891.
Até o presente momento, não há nenhum método analítico oficial descrito pela Farmacopeia Brasileira ou pelos compêndios internacionais para o controle de qualidade de formulações de CS. Na literatura são reportados vários trabalhos para análise de CS em amostras farmacêuticas por métodos cromatográficos, principalmente, por Cromatografia Líquida de Alta Eficiência (HPLC, High Performance Liquid Chromatography).33 Cooper, J. D. H.; Muirhead, D. C.; Taylor, J. E.; Baker, P. R.; J. Chromatogr. B: Biomed. Sci. Appl.
1997, 701, 87.
4 Dinesh, N. D.; Vishukumar, B. K.; Nagaraja, P.; Gowda, N. M. M.; Rangappa, K. S.; J. Pharm. Biomed. Anal.
2002, 29, 743.
5 Ortiz, R. S.; Antunes, M. V.; Linden, R.; Quim. Nova
2010, 33, 389.
6 Nickum, E. A.; Flurer, C. L.; J. Chromatogr. Sci.
2015, 53, 38.
7 Ghodsi, R.; Kobarfard, F.; Tabatabai, S. A.; Iran. J. Pharm. Res.
2012, 11, 123.-88 Fidan, A. K.; Bakirdere, S.; J. AOAC Int.
2016, 99, 923. Apesar da robustez desses métodos na determinação de SC em amostras farmacêuticas, o custo elevado das análises e o maior tempo requerido nestes protocolos são inconvenientes para um controle de qualidade de rotina nas indústrias farmacêuticas. Além disso, esses métodos cromatográficos utilizam solventes orgânicos na composição da fase móvel, gerando grandes quantidades de resíduos tóxicos. Neste sentido, na literatura são descritos outros métodos que buscam contornar esses problemas para determinação de CS em medicamentos, como por exemplo, utilizando a eletroforese capilar99 Vidal, D. T. R.; Augelli, M. A.; do Lago, C. L.; Anal. Methods
2013, 5, 2041., espectrofotometria UV-VIS,1010 Dinesh, N. D.; Nagaraja, P.; Gowda, N. M. M.; Rangappa, K. S.; Talanta
2002, 57, 757.
11 Amin, A. S.; Moustafa, M. E.; El-Dosoky, R. M. S.; J. AOAC Int.
2009, 92, 125.
12 Altiokka, G.; Atkosar, Z.; Sener, E.; Tuncel, M.; J. Pharm. Biomed. Anal.
2001, 25, 339.
13 Los Weinert, P.; Pezza, L.; Pezza, H. R.; Quim. Nova
2008, 31, 1112.
14 Issa, Y. M.; El-Hawary, W. F.; Youssef, A. F. A.; Senosy, A. R.; Spectrochim. Acta, Part A
2010, 75, 1297.-1515 Harikrishna, K.; Nagaralli, B. S.; Seetharamappa, J.; J. Food Drug Anal.
2008, 16, 11. dispersão de Rayleigh1616 Liu, S. P.; Fan, L.; Hu, X. L.; Liu, Z. F.; Li, S.; Anal. Sci.
2006, 22, 819. e, em destaque, o eletroanalítico devido à sua alta sensibilidade, seletividade e baixo custo de instrumentação.1717 Berzas, J. J.; Rodriguez, J.; Castaneda, G.; Villasenor, M. J.; Anal. Chim. Acta
2000, 417, 143.
18 Sopha, H.; Hocevar, S. B.; Pihlar, B.; Ogorevc, B.; Electrochim. Acta
2012, 60, 274.
19 Tyszczuk, K.; Korolczuk, M.; Bioelectrochemistry
2010, 78, 113.
20 Ozkan, S. A.; Uslu, B.; Zuman, P.; Anal. Chim. Acta
2004, 501, 227.
21 Baranowska, I.; Koper, M.; Markowski, P.; Chem. Anal.
2008, 53, 967.
22 Stefan-van Staden, R. I.; Van Staden, J. F.; Aboul-Enein, H. Y.; J. Solid State Electrochem.
2010, 14, 997.
23 Balasoiu, S. C.; Stefan-van Staden, R. I.; van Staden, J. F.; Ion, R. M.; Radu, G. L.; Aboul-Enein, H. Y.; Electrochim. Acta
2011, 58, 290.
24 Li, Y.; Wen, T. T.; Xue, C.; Han, Q.; Wang, Y.; Hong, J. L.; Zhou, X. M.; Jiang, H.; J. Biosens. Bioelectron.
2013, 42, 287.
25 Batista, E. F.; Sartori, E. R.; Medeiros, R. A.; Rocha, R. C.; Fatibello, O.; Anal. Lett.
2010, 43, 1046.
26 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci.
2012, 7, 13008.
27 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci.
2015, 10, 1494.-2828 Lopes, A. C. V.; Luz, R. D. S.; Damos, F. S.; dos Santos, A. S.; Franco, D. L.; dos Santos, W. T. P.; J. Braz. Chem. Soc.
2012, 23, 1800. Dentre esses métodos eletroanalíticos para a determinação de CS, as técnicas voltamétricas de pulso diferencial e onda quadrada são as mais aplicadas usando diversos eletrodos de trabalho, como por exemplo: gota pendente de mercúrio,1717 Berzas, J. J.; Rodriguez, J.; Castaneda, G.; Villasenor, M. J.; Anal. Chim. Acta
2000, 417, 143. filme de bismuto,1818 Sopha, H.; Hocevar, S. B.; Pihlar, B.; Ogorevc, B.; Electrochim. Acta
2012, 60, 274. filme de chumbo,1919 Tyszczuk, K.; Korolczuk, M.; Bioelectrochemistry
2010, 78, 113. carbono vítreo,2020 Ozkan, S. A.; Uslu, B.; Zuman, P.; Anal. Chim. Acta
2004, 501, 227., 2121 Baranowska, I.; Koper, M.; Markowski, P.; Chem. Anal.
2008, 53, 967. pasta de diamante,2222 Stefan-van Staden, R. I.; Van Staden, J. F.; Aboul-Enein, H. Y.; J. Solid State Electrochem.
2010, 14, 997. pasta de carbono modificada,2323 Balasoiu, S. C.; Stefan-van Staden, R. I.; van Staden, J. F.; Ion, R. M.; Radu, G. L.; Aboul-Enein, H. Y.; Electrochim. Acta
2011, 58, 290. carbono vítreo modificado com óxido de grafeno,2424 Li, Y.; Wen, T. T.; Xue, C.; Han, Q.; Wang, Y.; Hong, J. L.; Zhou, X. M.; Jiang, H.; J. Biosens. Bioelectron.
2013, 42, 287. diamante dopado com boro (BDD, Boron-Doped Diamond)2525 Batista, E. F.; Sartori, E. R.; Medeiros, R. A.; Rocha, R. C.; Fatibello, O.; Anal. Lett.
2010, 43, 1046. e eletrodos impressos (SPE, Screen- Printed Electrode).2626 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci.
2012, 7, 13008., 2727 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci.
2015, 10, 1494. Apesar do sucesso destes métodos, a maioria dos eletrodos sólidos utilizados nesses trabalhos está sujeita à passivação (envenenamento) devido à adsorção de espécies na superfície desses materiais. Na prática, este problema pode ser corrigido mediante o polimento e tratamento eletroquímico entre as medidas ou substituição do eletrodo após cada análise, como no caso dos eletrodos descartáveis.
Outra alternativa para diminuir a adsorção das espécies consiste em reduzir o tempo de contato entre o analito e a superfície do eletrodo de trabalho. Esta possibilidade pode ser alcançada utilizando sistemas hidrodinâmicos, como na Análise por Injeção em Fluxo (FIA, Flow Injection Analysis) ou Análise por Injeção em Batelada (BIA, Batch Injection Analysis). Nesses sistemas a técnica de detecção eletroquímica mais utilizada é a amperometria. Neste contexto, o CS foi determinado em formulações farmacêuticas por Lopes et al.2828 Lopes, A. C. V.; Luz, R. D. S.; Damos, F. S.; dos Santos, A. S.; Franco, D. L.; dos Santos, W. T. P.; J. Braz. Chem. Soc. 2012, 23, 1800. empregando um eletrodo de BDD em sistema FIA com detecção amperométrica de múltiplos pulsos. Vários outros fármacos também foram determinados por FIA ou BIA com detecção amperométrica, mostrando o grande potencial de aplicação desses métodos hidrodinâmicos.2929 Felix, F. S.; Angnes, L.; J. Pharm. Sci. 2010, 99, 4784., 3030 Quintino, M. S. M.; Angnes, L.; Electroanalysis 2004, 16, 513.
O sistema FIA é uma técnica que foi proposta em 1975 por Ruzicka e Hansen, na qual a solução contendo o analito é injetada durante o percurso de um fluxo com uma solução carregadora, que é movida por um sistema propulsor até o detector. 3131 Ruzicka, J.; Hansen, E. H.; Anal. Chim. Acta 1975, 78, 145. Já na técnica BIA, proposta em 1991 por Wang e Taha, um injetor introduz a solução do analito diretamente sobre a superfície de um detector, que está localizado em um recipiente (célula BIA) contendo uma solução inerte estacionária ou sob convecção (na presença de agitação magnética ou mecânica).3232 Wang, J.; Taha, Z.; Anal.Chem. 1991, 63, 1053. De acordo com a literatura, ambos os sistemas apresentam similaridade na proposta de análises realizadas continuamente e com menor intervenção do analista, apresentando picos transientes com alta frequência analítica, precisão e baixo consumo de amostras e reagentes. Embora o sistema BIA pode ser mais prático e portátil, o sistema FIA é mais versátil, uma vez que permite a adição de etapas no percurso antes da detecção do analito, como o tratamento da amostra, processo de separação, reações e/ou aquecimento.3030 Quintino, M. S. M.; Angnes, L.; Electroanalysis 2004, 16, 513. Dessa forma, como FIA e BIA oferecem vantagens para aplicação de métodos analíticos, faz-se necessário uma avaliação de qual sistema é o mais adequado para cada analito.
No cenário atual, os SPE descartáveis têm sido bastante utilizados para a eletroanálise de vários fármacos, incluindo CS.2626 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci. 2012, 7, 13008., 2727 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci. 2015, 10, 1494. Em uma recente revisão de Couto et al.3333 Couto, R. A. S.; Lima, J.; Quinaz, M. B.; Talanta 2016, 146, 801. é destacado o uso de SPE para a determinação individual e simultânea de aproximadamente cinquenta fármacos em medicamentos e matrizes biológicas. De modo geral, o uso destes SPE é atraente, pois são dispositivos práticos e portáteis que permitem realizar medidas em apenas uma gota de solução. Além disso, os SPE evitam a tediosa etapa de polimento do eletrodo (tal como ocorre em outros eletrodos sólidos), pois caso ocorra a passivação da superfície do seu eletrodo de trabalho, um novo SPE pode ser utilizado. Todavia, devido ao custo relativamente alto dos SPE (sobretudo os comerciais), a sua substituição após um limitado número de medidas pode tornar o seu uso inviável em análises de rotina. Como já mencionado, pode-se reduzir a passivação do eletrodo e aumentar a vida útil desses dispositivos acoplando o SPE em células eletroquímicas de análise por injeção em fluxo (FIA-SPE) ou em batelada (BIA-SPE).
O primeiro trabalho empregando FIA-SPE foi publicado em 1998, no qual os autores utilizaram reatores enzimáticos em linha com o SPE para determinar o frescor de peixes.3434 Carsol, M. A.; Mascini, M.; Talanta
1998, 47, 335. O trabalho pioneiro utilizando BIA-SPE é mais recente (2014), no qual foi proposto um sistema inteiramente portátil, adequado para análises em campo de modo prático, robusto e reprodutível.3535 Tormin, T. F.; Cunha, R. R.; da Silva, R. A. B.; Munoz, R. A. A.; Richter, E. M.; Sens. Actuators, B
2014, 202, 93. Após este trabalho, o mesmo grupo de pesquisa produziu, em parceria com uma empresa, uma versão comercial do sistema BIA-SPE.3636 Richter, E. M.; Tormin, T. F.; Cunha, R. R.; Silva, W. P.; Perez-Junquera, A.; Fanjul-Bolado, P.; Hernandez-Santos, D.; Munoz, R. A. A.; Electroanalysis
2016, 28, 1856. Atualmente, esses sistemas de detecção são comercializados em kits para a montagem dos sistemas FIA-SPE (célula, potenciostato portátil, bomba peristáltica, tubulações e injetor) e BIA-SPE (célula, potenciostato portátil e pipeta eletrônica).3737
http://www.dropsens.com/en/accesories_pag.html, acessada em 06/04/2017
http://www.dropsens.com/en/accesories_pa...
De modo alternativo, células eletroquímicas hidrodinâmicas para SPE de custo muito menor podem ser confeccionadas em laboratório com o uso de microrretífica,3838 Caramit, R. P.; Lucca, B. G.; Ferreira, V. S.; Munoz, R. A. A.; Richter, E. M.; da Silva, R. A. B.; Electroanalysis
2015, 27, 271. tornearia3535 Tormin, T. F.; Cunha, R. R.; da Silva, R. A. B.; Munoz, R. A. A.; Richter, E. M.; Sens. Actuators, B
2014, 202, 93. ou impressão 3D.3939 Dias, A. A.; Cardoso, T. M. G.; Cardoso, R. M.; Duarte, L. C.; Munoz, R. A. A.; Richter, E. M.; Coltro, W. K. T.; Sens. Actuators, B
2016, 226, 196. Apesar da praticidade e rapidez destes sistemas hidrodinâmicos com SPE, existem poucos trabalhos na literatura para determinação de fármacos em medicamentos por FIA-SPE4040 Bergamini, M. F.; Santos, A. L.; Stradiotto, N. R.; Zanoni, M. V. B.; J. Pharm. Biomed. Anal.
2007, 43, 315.
41 Fanjul-Bolado, P.; Lamas-Ardisana, P. J.; Hernandez-Santos, D.; Costa-Garcia, A.; Anal. Chim. Acta
2009, 638, 133.
42 Stefano, J. S.; Montes, R. H. O.; Richter, E. M.; Munoz, R. A. A.; J. Braz. Chem. Soc.
2014, 25, 484.-4343 Stefano, J. S.; Cordeiro, D. S.; Marra, M. C.; Richter, E. M.; Munoz, R. A. A.; Electroanalysis
2016, 28, 350. e BIA-SPE.4343 Stefano, J. S.; Cordeiro, D. S.; Marra, M. C.; Richter, E. M.; Munoz, R. A. A.; Electroanalysis
2016, 28, 350.
Dessa forma, este trabalho propõe a determinação de CS empregando SPE em sistemas BIA e FIA com detecção amperométrica, sendo avaliados cinco SPE de diferentes eletrodos de trabalho. A potencialidade e comparação do desempenho desses dois sistemas (BIA-SPE e FIA-SPE) para determinação de CS em formulações farmacêuticas foram avaliadas em função dos parâmetros analíticos.
PARTE EXPERIMENTAL
Reagentes e soluções
Todos os reagentes foram de grau analítico. O padrão de citrato de sildenafila (CS) foi obtido da Sigma-Aldrich®. As soluções foram preparadas com água deionizada obtida do sistema de purificação GEHAKA OS10LXE (resistividade > 18,2 MΩ cm). O tampão Britton-Robinson (TBR) 0,040 mol L-1 foi preparado com ácido acético, ácido bórico e ácido fosfórico, todos da Dinâmica (São Paulo, Brasil) e o ajuste do pH desejado foi realizado com solução de hidróxido de sódio (Vetec) e/ou ácido clorídrico, ambos a 0,2 mol L-1. O CS foi quantificado nos medicamentos da marca referência, Viagra (Pfizer®), e de quatro formulações genéricas (Eurofarma®, EMS®, Medley® e Neo Química®), todas com dosagem rotulada de 50 mg. Os comprimidos farmacêuticos foram adquiridos em drogarias da cidade de Dourados-MS.
As soluções estoque de CS foram preparadas em balão volumétrico, dissolvendo uma massa adequada do padrão de CS em água deionizada com auxílio de banho ultrassônico por 10 minutos. O mesmo procedimento foi adotado na preparação dos medicamentos de CS, porém o pó foi obtido triturando-se 20 comprimidos de cada medicamento (do mesmo lote). As soluções injetadas nos sistemas BIA e FIA foram preparadas no início do dia de trabalho, diluindo-se apropriadamente a solução estoque do padrão (ou amostra) no eletrólito suporte. Nas análises cromatográficas as soluções estoque de CS foram preparadas e diluídas em metanol (grau HPLC), sendo estas injetadas neste sistema após uma filtração.
Medidas Eletroquímicas
As medidas eletroquímicas foram realizadas com potenciostato portátil Dropsens® µStat400 interfaceado a um computador contendo o software Dropview8400®. A voltametria foi utilizada nos estudos do comportamento eletroquímico em diferentes eletrodos de trabalho e eletrólitos. A amperometria foi utilizada para detecção e determinação de CS nos sistemas hidrodinâmicos BIA e FIA. Para a detecção de CS foram utilizados os SPE comerciais da Dropsens®.3737
http://www.dropsens.com/en/accesories_pag.html, acessada em 06/04/2017
http://www.dropsens.com/en/accesories_pa...
Esses dispositivos contém os três eletrodos no mesmo substrato de cerâmica, sendo o eletrodo auxiliar de grafite e o eletrodo de pseudo-referência (EPR) de prata. Foram avaliados cinco SPE por voltametria, cada um contendo um eletrodo de trabalho distinto, sendo constituídos de: Carbono (grafite) não modificado (SPE-C); Carbono modificado com nanotubos de parede simples (SPE-SWCNT), nanotubos de parede múltipla (SPE-MWCNT), Grafeno (SPE-Gr) e Polianilina (SPE-PA). A área geométrica de todos os eletrodos de trabalho foi de 0,13 cm² (r = 2 mm). Nas medidas voltamétricas estacionárias, o SPE foi inserido em um béquer de 10 mL contendo o eletrólito suporte. Nas medidas amperométricas hidrodinâmicas, o melhor SPE selecionado para determinação de CS foi acomodado na mesma célula eletroquímica construída no laboratório e adaptada ora para o sistema BIA, ora para o sistema FIA.
Sistemas BIA-SPE e FIA-SPE
Na Fig. 1 é apresentado um esquema da célula eletroquímica hidrodinâmica, de configuração do tipo Wall Jet, que foi usada para os sistemas BIA-SPE e FIA-SPE. Nesta célula, o SPE é colocado em um orifício na parte inferior, contendo um anel cilíndrico de borracha (o-ring) por cima dos seus eletrodos, sendo este conjunto (SPE com o-ring) fixado por uma chapa rígida com dois parafusos. A tampa contém um orifício concêntrico ao eletrodo de trabalho do SPE, que é adequado para posicionar uma ponteira (Combitip®) de 1,0 mL da pipeta eletrônica. Assim, a extremidade inferior da Combitip possui distância sempre fixa de aproximadamente 2 mm da saída da solução até a superfície do eletrodo de trabalho no SPE, como já reportado.3030 Quintino, M. S. M.; Angnes, L.; Electroanalysis 2004, 16, 513. Esta célula com SPE é usada tanto em BIA quanto em FIA, porém cada sistema possui seus componentes específicos. Em BIA a injeção dos padrões e amostras é realizada por um injetor manual ou eletrônico (mais reprodutível), sendo que neste trabalho as soluções de CS foram injetadas por uma pipeta eletrônica Eppendorf® Multipette Stream. No sistema BIA, após a injeção da solução, a diluição do analito no eletrólito é acelerada por meio da convecção do eletrólito suporte.3232 Wang, J.; Taha, Z.; Anal.Chem. 1991, 63, 1053. Neste trabalho foi utilizado um agitador mecânico construído no próprio laboratório, o qual foi colocado na parte superior da célula ao lado do orifício da ponteira (detalhe em azul na Fig 1). No sistema FIA é necessário uma bomba peristáltica para transportar a solução do eletrólito no interior de tubos de polipropileno em direção ao SPE, sendo as soluções do analito inseridas no percurso por um injetor manual (ou automático). O sistema FIA utilizado neste trabalho foi de linha única e composto basicamente por tubos de polietileno de 1,0 mm de diâmetro interno, bomba peristáltica (Gilson®, modelo Minipuls 3) e um injetor manual comutador. Para manter a mesma distância da injeção no sistema BIA, a parte final do tubo de polietileno usado em FIA é inserida dentro da Combitip (detalhe em vermelho na Fig 1), com a solução saindo exatamente na extremidade inferior desta ponteira. Deste modo, a distância entre a extremidade inferior da ponteira e o SPE, bem como o diâmetro do jato de solução são sempre muito próximos em ambos os sistemas (FIA e BIA), garantindo uma comparação de resultados mais adequada.
Esquema com os principais componentes da célula eletroquímica Wall Jet para os sistemas: BIA-SPE com suas partes específicas (em azul) do agitador mecânico e da base de encaixe da pipeta eletrônica; e FIA-SPE com as suas partes específicas (em vermelho) do tubo de polietileno inserido dentro da ponteira, cuja solução é bombeada por uma bomba peristáltica e a injeção realizada por um injetor manual. Em destaque a região ampliada do encaixe do SPE na célula e de cada eletrodo no SPE.
A célula eletroquímica construída possui disposição semelhante ao primeiro sistema BIA-SPE proposto e dispensa o uso de suporte,3535 Tormin, T. F.; Cunha, R. R.; da Silva, R. A. B.; Munoz, R. A. A.; Richter, E. M.; Sens. Actuators, B 2014, 202, 93. permitindo o fácil posicionamento em bancadas de laboratório ou em qualquer superfície horizontal. Além disso, o material do corpo da célula é de policloreto de vinila (PVC), um polímero com alta resistência mecânica e química. Imagens e outros detalhes desta célula para os sistemas BIA e FIA podem ser consultadas no material suplementar.
Análises por Cromatografia Líquida de Alta Eficiência (HPLC)
Os medicamentos contendo CS foram também analisados por HPLC para a comparação dos métodos propostos. Os experimentos foram realizados em um cromatógrafo líquido da Shimadzu (Modelo Prominence LC 20 AD) com detector no UV com arranjo de diodos (SPD-M20A), injetor automático (SIL-10AF), bomba de pistão LC-6Adx3 e coluna C18 Shim-Pack XROD de 15 mm de comprimento e 2 mm de diâmetro interno, com temperatura controlada (forno CTO-20A) em 40 ºC. Os experimentos por HPLC foram conduzidos no modo isocrático com fase móvel contendo 100 % de metanol. Deste modo, o CS apresentou um tempo de retenção de 2,16 min e foi quantificado nas cinco amostras farmacêuticas em 290 nm. As condições cromatográficas foram adaptadas de um trabalho prévio para a análise de CS.2828 Lopes, A. C. V.; Luz, R. D. S.; Damos, F. S.; dos Santos, A. S.; Franco, D. L.; dos Santos, W. T. P.; J. Braz. Chem. Soc. 2012, 23, 1800. Os resultados obtidos por FIA-SPE e BIA-SPE para o doseamento, em triplicata, do CS nos cinco medicamentos foram comparados com HPLC-UV aplicando os testes estáticos t e F, ambos com nível de confiança de 95%.
RESULTADOS E DISCUSSÃO
O comportamento eletroquímico do CS foi avaliado sob SPE-C por voltametria cíclica em meio de soluções tampão Britton-Robinson (TBR) 0,04 mol L-1 em pH de 2 a 10. Os resultados são apresentados na Figura 2, sendo possível observar processos de oxidação irreversíveis para CS, cujos potenciais de pico se tornam menos positivos com o incremento de pH. No voltamograma do pH 2 (Figura 2), o CS apresentou apenas um processo de oxidação nítido em torno de 1,1 V, porém de pH 4 a 7 foram observados dois processos de oxidação, os quais a partir de pH 8 foram tornando-se menos perceptíveis nos voltamogramas. Estes resultados corroboram com o perfil de CS observado em eletrodos de Carbono Vítreo2020 Ozkan, S. A.; Uslu, B.; Zuman, P.; Anal. Chim. Acta 2004, 501, 227. e BDD,2525 Batista, E. F.; Sartori, E. R.; Medeiros, R. A.; Rocha, R. C.; Fatibello, O.; Anal. Lett. 2010, 43, 1046., 2828 Lopes, A. C. V.; Luz, R. D. S.; Damos, F. S.; dos Santos, A. S.; Franco, D. L.; dos Santos, W. T. P.; J. Braz. Chem. Soc. 2012, 23, 1800. Na literatura, um mecanismo sugerido é de que o primeiro processo anódico é correspondente à oxidação do anel piperazínico da molécula de sildenafila e o segundo é devido à adsorção/dessorção do produto gerado no primeiro.2020 Ozkan, S. A.; Uslu, B.; Zuman, P.; Anal. Chim. Acta 2004, 501, 227. Para o SPE-C não foi observado o segundo pico anódico em pH 2, provavelmente devido à sobreposição dos dois picos neste valor de pH. Como a corrente de pico de oxidação para o CS foi mais sensível em pH 2 no TBR 0,04 mol L-1, este eletrólito foi escolhido nas medidas subsequentes. Apesar do deslocamento dos potenciais de pico do CS em valores mais altos de pH proporcionarem uma melhor seletividade para detecção deste analito, esses pHs não foram escolhidos pelo fato destes medicamentos não apresentarem interferentes para detecção eletroquímica nesta faixa de potencial.
Voltamogramas cíclicos obtidos sob SPE-C em meio de TBR 0,04 mol L-1 (pH de 2 a 10) sem e com a adição de CS 75 µmol L-1. Velocidade de varredura: 50 mV s-1
Diferentes tipos de eletrodos impressos foram avaliados para detecção de CS em função da melhor sensibilidade e repetibilidade das respostas eletroquímica. Este estudo foi realizado para dez medidas sucessivas de solução de CS 5,0 µmol L-1 em cada SPE por voltametria de onda quadrada. Os parâmetros da técnica (frequência, amplitude e incremento de potencial) foram otimizados adotando como critério a melhor sensibilidade para detecção do CS. Na Fig. 3A é apresentado o primeiro voltamograma obtido para cada eletrodo impresso avaliado: Carbono não modificado (SPE-C) e modificado com nanotubo de parede simples (SPE-SWCNT), nanotubo de parede múltipla (SPE-MWCNT), Grafeno (SPE-Gr) e Polianilina (SPE-PA). Com exceção do SPE-Gr que apresentou o maior potencial de pico anódico (Epa) para a oxidação de sildenafila, os demais SPE proporcionaram um Epa semelhante para este composto, em aproximadamente +1,05 V. Deste modo, os SPE modificados não promoveram nenhum efeito catalítico para o CS. Na Fig. 3A, observa-se também que a corrente de fundo obtida no SPE-Gr (> 200 µA) foi muito superior em relação aos outros SPE. Este comportamento do SPE-Gr pode ser justificado pelo ordenamento tridimensional do grafeno, o qual é responsável por controlar a capacitância do eletrodo. De acordo com a literatura, 4444 Ke, Q.; Wang, J.; J. Materiomics 2016, 2, 37. em muitos casos as unidades de grafeno estão irregularmente empilhadas devido às aglomerações não controladas durante a síntese ou uso deste eletrodo, podendo haver um aumento da corrente capacitiva. Além disso, a fragilidade da superfície do SPE-Grafeno da Dropsens® foi observada pelo constante desprendimento de grafeno do eletrodo de trabalho, antes e durante as análises, o que provoca ainda mais variações na área eletroquimicamente ativa deste eletrodo e, consequentemente, pode ter aumentado a sua corrente capacitiva.
(A) Voltamogramas de onda quadrada da primeira varredura ob tidos em solução de CS 5,0 µmol L-1 nos distintos SPE: C, MWCNT, SWCNT, Gr e PA; (B) Correntes de pico médias (± DP) obtidas para dez varreduras sucessivas de CS em cada SPE. Eletrólito: TBR 0,04 mol L-1 pH 2; Frequência: 20 Hz; Amplitude: 25 mV, Incremento de Potencial: 3 mV
Na Figura 3B são apresentados os valores de correntes de pico médias (± DP) de dez voltamogramas de onda quadrada sucessivos para cada SPE. Nesta Figura 3B é possível observar, com exceção do SPE-SWCNT, que os eletrodos modificados apresentaram correntes de pico (ipa) levemente superiores ao eletrodo não modificado (SPE-C). Entretanto, a repetibilidade desses sinais nos 10 voltamogamas para detecção do CS foi muito baixa para o SPE-Gr e SPE-PA, com desvio padrão relativo (DPR) próximo a 30 % nesses eletrodos. Os sensores que apresentaram maior precisão foram SPE-C e SPE-MWCNT (DPR ≈ 9 % para ambos), sendo a ipa média apenas de 10 % menor para o SPE-C. Deste modo, o eletrodo impresso de carbono não modificado (SPE-C) foi escolhido para as medidas amperométricas nos sistemas BIA e FIA, uma vez que este dispositivo apresenta um custo de aquisição em torno de 60 % mais baixo do que o SPE-MWCNT.
Para a investigação da resposta eletroquímica da sildenafila nos sistemas hidrodinâmicos com detecção amperométrica, uma solução de CS 5,0 µmol L-1 foi injetada em triplicata na célula BIA preenchida com solução TBR 0,04 mol L-1 pH 2. A detecção amperométrica foi realizada aplicando um potencial constante no SPE-C em função do tempo, sendo avaliados os potenciais aplicados de 0,0 V até + 1,2 V (vs. EPR). O voltamograma hidrodinâmico apresentado na Figura 4 foi construído a partir dos valores médios (± DP) das correntes de pico anódicas em cada potencial aplicado. Na Figura 4 pode ser observado que o início da oxidação do CS na superfície do SPE-C ocorre a partir de + 0,9 V e alcança um valor máximo em + 1,2 V. Este comportamento corrobora com o observado por voltametria cíclica no mesmo eletrólito (Figura 2). Embora a maior corrente de pico anódica (ipa) ter sido obtida em + 1,2 V, neste potencial a corrente de fundo (linha de base) foi mais intensa e instável, provavelmente, devido à descarga da água na superfície do SPE-C. Assim, o CS foi detectado no potencial de + 1,1 V no SPE-C nos estudos seguintes para os sistemas BIA e FIA.
Voltamogramas hidrodinâmicos obtidos plotando as correntes de pico médias (± DP) para três injeções da solução de CS 5,0 µmol L-1 no sistema BIA com detecção amperométrica em potenciais distintos. Eletrólito: TBR 0,04 mol L-1 pH 2; Volume injetado: 100 µL; Vazão: 134 µL s-1
Os parâmetros de injeção (volume e a vazão) nos sistemas BIA e FIA foram investigados para obter as melhores respostas analíticas em função da sensibilidade e frequência analítica. Os estudos foram realizados injetando uma solução de CS 5,0 µmol L-1, em triplicada, sob cada condição de modo univariado, ou seja, variando o volume injetado sob uma vazão fixa e, posteriormente, variando a vazão sob um volume fixo. No estudo do volume injetado no sistema BIA, a pipeta eletrônica (Eppendorf®) acoplada na ponteira de 1,0 mL permitiu a injeção de volumes no intervalo de 10 a 1000 µL. As correntes médias (± DP) para três injeções deste estudo são apresentadas na Fig. 5A (BIA). Neste estudo observa-se que quando são injetados volumes de 10 a 100 µL a corrente de pico é diretamente proporcional ao volume injetado na solução. No entanto, acima de 100 µL estes sinais são praticamente constantes e os desvios são maiores, provavelmente, devido à maior passivação da superfície do eletrodo de trabalho. Além disso, a frequência analítica é menor em volumes mais altos de injeção, obtendo-se picos mais largos. Deste modo, o volume de 50 µL foi escolhido para a injeção no sistema BIA. Já no sistema FIA, a magnitude do volume injetado no percurso da solução carregadora (eletrólito suporte) depende da alça de amostragem ("looping") que é utilizada no injetor comutador. Como também apresentado na Fig. 5A (FIA), as alturas de correntes são diretamente proporcionais aos volumes injetados em todo o intervalo avaliado (75 até 250 µL), sendo a corrente máxima obtida no volume de 250 µL. Este comportamento é observado no FIA devido à dispersão longitudinal do CS no eletrólito durante o percurso analítico (distância do injetor até o SPE). Em outras palavras, a concentração no centro do volume injetado é menor quando alcança o SPE em relação à concentração inicial no injetor. Esse efeito só é eliminado em grandes volumes de injeção, pois as alturas das correntes passam a permanecer constante com aumento do volume injetado. No entanto, alças de amostragem acima de 250 µL não foram avaliadas devido à diminuição da frequência analítica e possibilidade de maior contaminação da superfície do eletrodo de trabalho. Dessa forma, a escolha da alça de amostragem foi baseada na maior precisão da resposta eletroquímica no sistema FIA-SPE, sendo escolhido o volume injetado de 150 µL para os experimentos posteriores.
Correntes de pico médias e respectivos desvios (ipa ± DP) para três injeções de solução de CS 5,0 µmol L-1 nos sistemas BIA e FIA em SPE-C, variando os volumes injetados em (A), vazões em (B) e agitação mecânica do eletrólito em BIA em (C). Condições fixadas em: (A) Vazão de 134 µL s-1 (BIA) e 66 µL s-1 (FIA); (B) Volume injetado de 50 µL (BIA) e 75 µL (FIA); (C) Volume de 50 µL e Vazão de 100 µL s-1. Eletrólito: TBR 0,04 mol L-1 pH 2; Potencial: + 1,1 V (vs. EPR)
No sistema BIA, a pipeta eletrônica usada neste trabalho permite a injeção das soluções sob dez vazões diferentes (24 a 250 µL s-1 na ponteira de 1,0 mL). As correntes de pico médias das triplicatas (± DP) em cada vazão são apresentadas na Fig. 5B (BIA), sendo os sinais diretamente proporcionais às velocidades de injeção devido à diminuição da camada de difusão de Nernst. No entanto, no sistema BIA a precisão da coleta dos sinais de corrente foi reduzida em vazões acima de 150 µL s-1. Esse fenômeno pode ocorrer devido à velocidade de aquisição do sinal não ser suficiente para coletar as correntes do analito que são registradas no pico (patamar de corrente), uma vez que nessas condições o composto eletroativo pode passar muito rápido pela superfície do eletrodo. Assim, a vazão de 100 µL s-1 foi usada nas próximas injeções para o sistema BIA. No sistema FIA, a bomba peristáltica usada neste trabalho permitiu vazões de 8 a 75 µL s-1 com os tubos utilizados (diâmetro interno de 1,0 mm). Como mostrado na Fig. 5B (FIA) entre o intervalo avaliado de 8 a 75 µL s-1, a maior corrente de pico foi obtida na maior vazão do sistema. No caso do sistema FIA, como o volume injetado ainda tem um efeito de dispersão, a altura da corrente pode ser aumentada em vazões altas devido ao menor tempo disponível para a diluição da solução de CS na solução carregadora, reduzindo a dispersão no centro da zona de amostra. Além disso, há o aumento da corrente de pico devido à diminuição da camada de difusão de Nernst com incremento da vazão. Deste modo, as determinações de CS por FIA foram realizadas na vazão máxima de 75 µL s-1.
O último parâmetro investigado, apenas para o sistema BIA, foi em relação à agitação mecânica do eletrólito no interior da célula. Neste estudo, soluções de CS 5,0 µmol L-1 foram injetadas, em triplicata, mantendo a vazão e o volume constantes sem agitação. Na sequência, o agitador foi ligado na rotação inicial de 700 rpm e novas injeções da mesma solução, em triplicata, foram realizadas. Deste modo, prosseguiu-se o experimento realizando novo incremento na rotação seguido de novas triplicatas até o nível máximo de agitação (4000 rpm). O nível de agitação é proporcional à tensão aplicada pela fonte universal do motor DC (de 3 a 12 V) utilizado no sistema BIA. Como mostrado na Fig. 5C, agitações até 1500 rpm aumentam a precisão das medidas em relação à medida sem agitação. Além disso, a velocidade de análise na presença de agitação foi bastante superior do que na ausência de agitação (duração de um pico ≈ 10 s com agitação a 700 rpm vs. 40 s sem agitação). Empiricamente, este aumento da velocidade é percebido pelo decaimento mais rápido do pico, que pode ser justificado pela remoção acelerada das espécies eletroativas próximas à superfície do eletrodo de trabalho em função da convecção gerada pelo motor DC.3232 Wang, J.; Taha, Z.; Anal.Chem. 1991, 63, 1053. Entretanto, uma convecção acima de 1500 rpm causa grande diminuição na precisão e na corrente de pico, pois a convecção excessiva provoca uma turbulência na solução da célula, podendo remover parte da zona da solução de CS antes mesmo de sua total detecção no eletrodo de trabalho. Outro ponto importante da agitação é diminuir a contaminação do eletrodo de trabalho devido ao menor tempo de contato direto do analito com o sensor após a detecção.4343 Stefano, J. S.; Cordeiro, D. S.; Marra, M. C.; Richter, E. M.; Munoz, R. A. A.; Electroanalysis 2016, 28, 350. Assim, as determinações de CS foram realizadas com uma agitação branda, a 700 rpm.
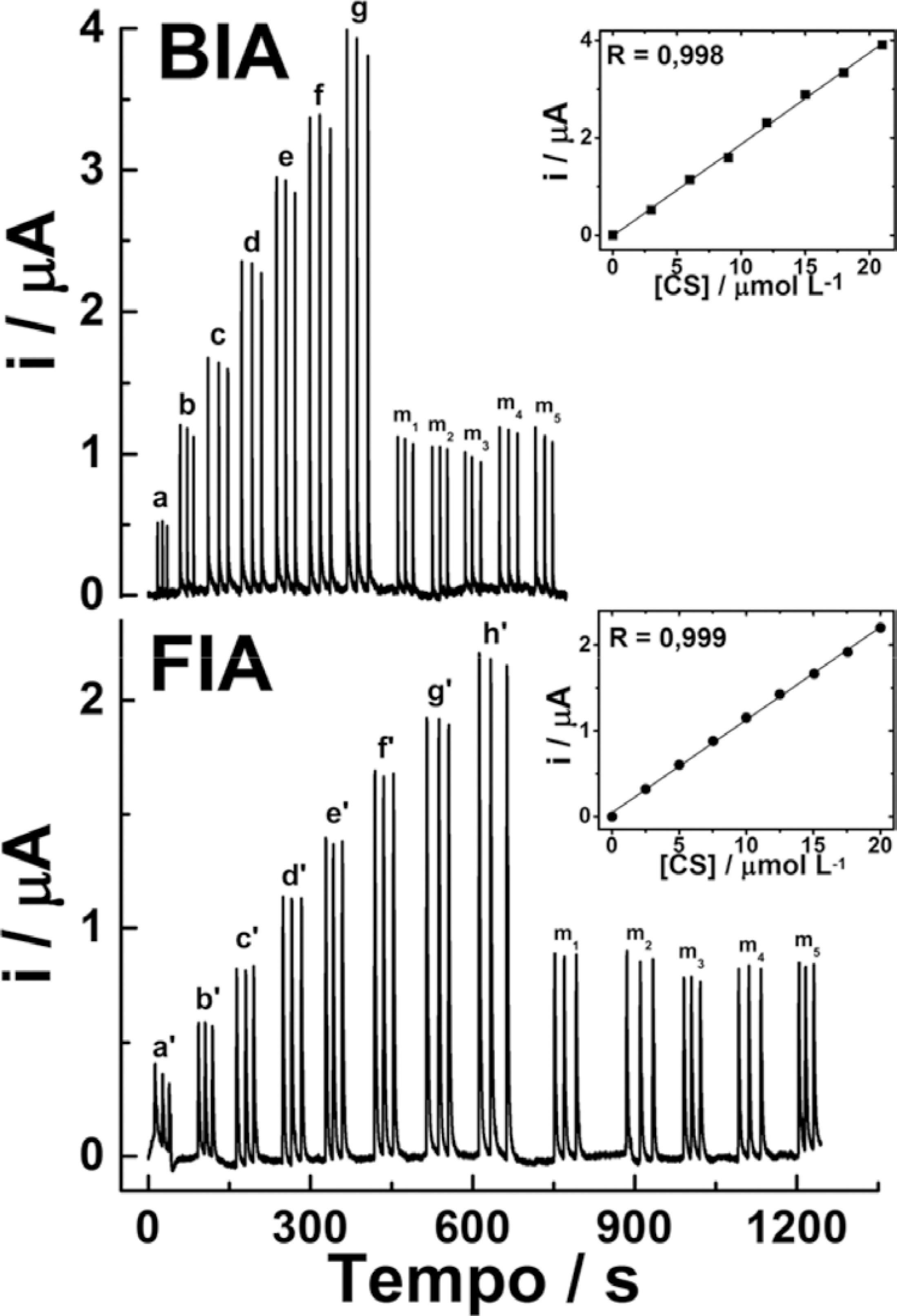
Após a otimização das condições nos sistemas hidrodinâmicos com detecção amperométrica, soluções de concentrações crescentes do padrão de CS foram injetadas em triplicata nos sistemas BIA e FIA. Deste modo, foi possível obter alguns parâmetros de confiabilidade analítica, como sensibilidade, faixa linear e limite de detecção (LD). Na sequência deste estudo, soluções diluídas dos cinco medicamentos foram injetadas em triplicata, sendo o CS quantificado em cada formulação por meio da interpolação das respectivas correntes de pico nas curvas analíticas de calibração (externa), as quais foram obtidas pelos amperogramas dos sistemas BIA-SPE e FIA-SPE. Na Figura 6 são apresentados os amperogramas para ambos os experimentos e as respectivas curvas de calibração. A partir destes resultados, foram calculadas as seguintes equações de reta: i(µA) = 0,188 [CS] (µmol L-1) - 0,0131 (R = 0,998) para o sistema BIA; e i(µA) = 0,108 [CS] (µmol L-1) + 0,0462 (R = 0,999) para o sistema FIA. Os LD estimados foram de 0,0520 µmol L-1 (BIA) e 0,215 µmol L-1 (FIA). As faixas lineares apresentadas em ambos os métodos foram estreitas (de 3,00 a 21,0 µmol L-1 para o sistema BIA e de 2,50 a 20,0 µmol L-1 para FIA), sendo essas semelhantes às faixas observadas de outros trabalhos para a detecção de CS em eletrodos impressos,2626 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci. 2012, 7, 13008., 2727 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci. 2015, 10, 1494. carbono vítreo2121 Baranowska, I.; Koper, M.; Markowski, P.; Chem. Anal. 2008, 53, 967. e BDD.2525 Batista, E. F.; Sartori, E. R.; Medeiros, R. A.; Rocha, R. C.; Fatibello, O.; Anal. Lett. 2010, 43, 1046. Todos estes parâmetros analíticos são apresentados na Tabela 1.
Injeções em triplicata de solução padrão de CS no sistema BIA (3,0 - 21,0 µmol L-1, a - g) e sistema FIA (2,5 - 20 µmol L-1, a' - h'), bem como das soluções diluídas das cinco amostras de medicamentos de CS (m1 a m5) em ambos os sistemas. As inserções representam as respectivas cur vas de calibração de cada sistema. Volume injetado: 50 µL (BIA) e 150 µΛ (ΦIA); vazão: 100 µL s-1 (BIA) e 75 µL s-1 (FIA); eletrólito: TBR 0,04 mol L-1 pH 2; potencial aplicado: + 1,10 V (vs. EPR); agitação mecânica da célula BIA: 700 rpm.
Para estimar a precisão e a frequência analítica teórica (TAF, theoretical analytical frequency), foram realizadas três injeções alternadas de solução de CS a 2,0 µmol L-1 e 15,0 µmol L-1 nos sistemas BIA e FIA. Como apresentado nos amperogramas da Figura 7, nota-se que os DPR das correntes de pico para as injeções de CS 2,0 µmol L-1 são próximos tanto para o sistema BIA (DPR = 3,61 %; n = 15), quanto para o sistema FIA (DPR = 3,76 %; n = 15). O mesmo comportamento ocorreu nos DPR paras as correntes de picos referentes à solução de CS 15,0 µmol L-1 calculados em 3,02 e 3,20 % (n = 15) para os sistemas BIA e FIA, respectivamente. Estes resultados indicam que a passivação (e/ou contaminação) do SPE-C é mínima quando estes eletrodos são acoplados aos sistemas BIA e FIA. Cabe destacar que a repetibilidade obtida para SPE-C pela voltametria de onda quadrada em estado estacionário (Fig. 2B) foi muito baixa para 10 medidas de CS 5,0 µmol L-1 (DPR = 9,0 %). Essa diferença entre os DPRs obtidos para os métodos hidrodinâmicos propostos e a voltametria estacionária demonstra a grande potencialidade dos sistemas FIA e BIA para aumentar a estabilidade da detecção eletroquímica.
Amperogramas obtidos para injeções em triplicatas e alternadas nos sistemas BIA e FIA de solução de CS de 2,0, e 15,0 mmol L-1. Condições de cada sistema idem a Figura 6.
Os valores das TAF foram calculados obtendo a média das larguras de pico das injeções de CS 15 mmol L-1 (Figura 7), estimando o número de injeções por hora para cada sistema. Nos dois casos, as TAF foram calculadas considerando que as injeções são realizadas em sequência, desprezando o tempo para preencher a pipeta no sistema BIA e a alça de amostragem no sistema FIA. Os valores das TAF estão apresentados na Tabela 1.
Na Tabela 1 também pode ser comparado o desempenho das duas formas de injeção (BIA e FIA) com detecção amperométrica em função dos parâmetros analíticos apresentados para determinação de CS. Nesta Tabela observa-se que o sistema BIA foi o mais sensível, provavelmente, devido às maiores vazões permitidas pela pipeta eletrônica em relação à bomba peristáltica utilizada neste trabalho para o sistema FIA, bem como da menor dispersão do pacote da solução injetada em BIA.3030 Quintino, M. S. M.; Angnes, L.; Electroanalysis 2004, 16, 513. O fato da maior vazão proporcionada pela pipeta eletrônica também explica a maior frequência analítica obtida no sistema BIA. A estabilidade do sinal eletroquímico de ambos os sistemas foi semelhante para soluções concentradas e diluídas de CS (DPR ≈ 3 %), mostrando que cada SPE-C pode ser reutilizado para várias medidas de CS. Na literatura apenas duas publicações comparam o desempenho entre BIA e FIA com detecção amperométrica para um mesmo analito. Ferreira et al. determinaram o ciclopirox em eletrodo de carbono vítreo, 4545 Ferreira, L. M. C.; Felix, F. S.; Angnes, L.; Electroanalysis 2012, 24, 961. obtendo valores de TAF de 735 h-1 e 330 h-1 com sensibilidades de 0,091 e 0,022 (µA mmol-1 L) para os sistemas BIA e FIA, respectivamente. Stefano et al.4343 Stefano, J. S.; Cordeiro, D. S.; Marra, M. C.; Richter, E. M.; Munoz, R. A. A.; Electroanalysis 2016, 28, 350. empregaram um SPE modificado na eletroanálise de ciprofloxacina e alcançaram valores de TAF de 450 h-1 e 180 h-1 com sensibilidades de 0,1115 e 0,0768 (µA mmol-1 L) para os sistemas BIA e FIA, respectivamente. Deste modo, tal como observado no presente trabalho, as comparações na literatura entre os dois sistemas para a análise de outros compostos, empregando diferentes células e eletrodos, também exibiram uma maior sensibilidade e frequência analítica para o sistema BIA. Cabe destacar que, na Figura 7 é observado um nível de ruído maior na detecção amperométrica em FIA, sendo o provável motivo que elevou o valor de LD para este sistema quando comparado ao obtido pelo BIA. O ruído apresentado em FIA pode ser justificado pela pulsação da bomba peristáltica utilizada neste trabalho, podendo este efeito ser minimizado pela utilização de um amortecedor de pulso ou de um sistema de propulsão alternativo.4646 Dos Santos, W. T. P.; Ceolin, M. P.; de Albuquerque, Y. D. T.; Richter, E. M.; Quim. Nova, 2007, 30, 1754.
A eficiência dos métodos desenvolvidos por BIA e FIA com detecção amperométrica foi avaliada para a determinação de CS em cinco amostras de medicamentos comerciais (Viagra® e genéricos). Os doseamentos de CS obtidos nestas amostras tanto pelos métodos propostos quanto por HPLC-UV são apresentados na Tabela 2.
Comparação entre os resultados da determinação de CS em medicamentos pelos métodos propostos e por HPLC-UV
A precisão e exatidão dos resultados obtidos pelos dois métodos propostos para determinação de CS são semelhantes aos apresentados por HPLC-UV. No teste F, as razões das variâncias do conjunto das análises por cada técnica proposta em relação com HPLC (F = Sproposto2/SHPLC 2) foram de 1,43 e 1,28 para BIA e FIA respectivamente, sendo estes valores inferiores ao Ftabelado em um nível de confiança de 95 % (F = 2,48; a = 14). No teste t pareado, o tcalculado foi de 1,38 e 0,82 para BIA e FIA respectivamente, sendo ambos inferiores ao ttabelado também a um nível de confiança de 95 % (2,15; a = 14). Deste modo, os resultados comprovam que não há diferença significativa entre todos os dados obtidos. Assim, os resultados confirmam que os métodos de BIA e FIA com detecção amperométrica possuem desempenho similar ao HPLC para as determinações de CS em formulações farmacêuticas.
Comparando os parâmetros analíticos apresentados no presente trabalho (Tabela 1), foi observado que os limites de detecção obtidos pelos métodos BIA-SPE e FIA-SPE foram superiores a vários outros trabalhos da literatura para a determinação de CS com detecção eletroquímica.1818 Sopha, H.; Hocevar, S. B.; Pihlar, B.; Ogorevc, B.; Electrochim. Acta
2012, 60, 274.,1919 Tyszczuk, K.; Korolczuk, M.; Bioelectrochemistry
2010, 78, 113.,2121 Baranowska, I.; Koper, M.; Markowski, P.; Chem. Anal.
2008, 53, 967.
22 Stefan-van Staden, R. I.; Van Staden, J. F.; Aboul-Enein, H. Y.; J. Solid State Electrochem.
2010, 14, 997.
23 Balasoiu, S. C.; Stefan-van Staden, R. I.; van Staden, J. F.; Ion, R. M.; Radu, G. L.; Aboul-Enein, H. Y.; Electrochim. Acta
2011, 58, 290.
24 Li, Y.; Wen, T. T.; Xue, C.; Han, Q.; Wang, Y.; Hong, J. L.; Zhou, X. M.; Jiang, H.; J. Biosens. Bioelectron.
2013, 42, 287.
25 Batista, E. F.; Sartori, E. R.; Medeiros, R. A.; Rocha, R. C.; Fatibello, O.; Anal. Lett.
2010, 43, 1046.
26 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci.
2012, 7, 13008.
27 Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci.
2015, 10, 1494.-2828 Lopes, A. C. V.; Luz, R. D. S.; Damos, F. S.; dos Santos, A. S.; Franco, D. L.; dos Santos, W. T. P.; J. Braz. Chem. Soc.
2012, 23, 1800. Entretanto, esta característica não se trata de um inconveniente e nem resulta em prejuízos na análise, uma vez que os métodos BIA-SPE e FIA-SPE são propostos apenas para a determinação de CS em amostras de formulações farmacêuticas. Em contrapartida, os métodos hidrodinâmicos apresentados neste trabalho proporcionaram uma frequência analítica muito maior frente aos demais métodos reportados para determinação de CS, o que representa uma grande vantagem na aplicação em análises de rotina deste fármaco. Além disso, a praticidade dos sistemas BIA-SPE e FIA-SPE pode ser um grande atrativo destes métodos para aplicação no controle de qualidade do CS pela indústria farmacêutica.
CONCLUSÃO
Este trabalho demonstrou dois métodos simples e rápidos para a determinação de CS em formulações farmacêuticas empregando eletrodos impressos de carbono (grafite) acoplados aos sistemas FIA e BIA com detecção amperométrica. O sistema BIA proporcionou uma maior sensibilidade e frequência analítica com menor consumo de reagentes (eletrólito, padrão e amostra), além de oferecer um sistema mais simples e portátil para aplicação do método. Os dois métodos propostos para determinação de CS foram comparados com o método cromatográfico (HPLC-UV) e os resultados de doseamento do fármaco mostraram-se semelhantes. A praticidade e as elevadas frequências analíticas obtidas pelos sistemas FIA-SPE e BIA-SPE para determinação de CS em relação aos demais trabalhos descritos são grandes atrativos para análises de rotina na indústria farmacêutica, abrindo uma perspectiva para que os sistemas propostos sejam empregados para outros fármacos eletroativos.
AGRADECIMENTOS
Ao CNPq (475276/2013-2 e 481683/2013-5) e à FUNDECT-MS (23/200.498/2014) pelo suporte financeiro. Rafael S. Backes agradece à CAPES pela bolsa de mestrado concedida. Este trabalho é uma colaboração entre projetos de pesquisa de membros da Rede Mineira de Química (RQ-MG) apoiada pela FAPEMIG (Projeto: CEX-RED-00010-14).
MATERIAL SUPLEMENTAR
Maiores detalhes (imagens e especificações) dos componentes dos sistemas BIA-SPE e FIA-SPE utilizados neste trabalho estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
REFERÊNCIAS
-
1Terrett, N. K.; Bell, A. S.; Brown, D.; Ellis, P.; Bioorg. Med. Chem. Lett. 1996, 6, 1819.
-
2Hurtado, R. L.; Lasmar, M. C.; Cad. Saúde Pública 2014, 30, 891.
-
3Cooper, J. D. H.; Muirhead, D. C.; Taylor, J. E.; Baker, P. R.; J. Chromatogr. B: Biomed. Sci. Appl. 1997, 701, 87.
-
4Dinesh, N. D.; Vishukumar, B. K.; Nagaraja, P.; Gowda, N. M. M.; Rangappa, K. S.; J. Pharm. Biomed. Anal. 2002, 29, 743.
-
5Ortiz, R. S.; Antunes, M. V.; Linden, R.; Quim. Nova 2010, 33, 389.
-
6Nickum, E. A.; Flurer, C. L.; J. Chromatogr. Sci. 2015, 53, 38.
-
7Ghodsi, R.; Kobarfard, F.; Tabatabai, S. A.; Iran. J. Pharm. Res. 2012, 11, 123.
-
8Fidan, A. K.; Bakirdere, S.; J. AOAC Int. 2016, 99, 923.
-
9Vidal, D. T. R.; Augelli, M. A.; do Lago, C. L.; Anal. Methods 2013, 5, 2041.
-
10Dinesh, N. D.; Nagaraja, P.; Gowda, N. M. M.; Rangappa, K. S.; Talanta 2002, 57, 757.
-
11Amin, A. S.; Moustafa, M. E.; El-Dosoky, R. M. S.; J. AOAC Int. 2009, 92, 125.
-
12Altiokka, G.; Atkosar, Z.; Sener, E.; Tuncel, M.; J. Pharm. Biomed. Anal. 2001, 25, 339.
-
13Los Weinert, P.; Pezza, L.; Pezza, H. R.; Quim. Nova 2008, 31, 1112.
-
14Issa, Y. M.; El-Hawary, W. F.; Youssef, A. F. A.; Senosy, A. R.; Spectrochim. Acta, Part A 2010, 75, 1297.
-
15Harikrishna, K.; Nagaralli, B. S.; Seetharamappa, J.; J. Food Drug Anal. 2008, 16, 11.
-
16Liu, S. P.; Fan, L.; Hu, X. L.; Liu, Z. F.; Li, S.; Anal. Sci. 2006, 22, 819.
-
17Berzas, J. J.; Rodriguez, J.; Castaneda, G.; Villasenor, M. J.; Anal. Chim. Acta 2000, 417, 143.
-
18Sopha, H.; Hocevar, S. B.; Pihlar, B.; Ogorevc, B.; Electrochim. Acta 2012, 60, 274.
-
19Tyszczuk, K.; Korolczuk, M.; Bioelectrochemistry 2010, 78, 113.
-
20Ozkan, S. A.; Uslu, B.; Zuman, P.; Anal. Chim. Acta 2004, 501, 227.
-
21Baranowska, I.; Koper, M.; Markowski, P.; Chem. Anal. 2008, 53, 967.
-
22Stefan-van Staden, R. I.; Van Staden, J. F.; Aboul-Enein, H. Y.; J. Solid State Electrochem. 2010, 14, 997.
-
23Balasoiu, S. C.; Stefan-van Staden, R. I.; van Staden, J. F.; Ion, R. M.; Radu, G. L.; Aboul-Enein, H. Y.; Electrochim. Acta 2011, 58, 290.
-
24Li, Y.; Wen, T. T.; Xue, C.; Han, Q.; Wang, Y.; Hong, J. L.; Zhou, X. M.; Jiang, H.; J. Biosens. Bioelectron. 2013, 42, 287.
-
25Batista, E. F.; Sartori, E. R.; Medeiros, R. A.; Rocha, R. C.; Fatibello, O.; Anal. Lett. 2010, 43, 1046.
-
26Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci. 2012, 7, 13008.
-
27Farghali, R. A.; Ahmed, R. A.; Int. J. Electrochem. Sci. 2015, 10, 1494.
-
28Lopes, A. C. V.; Luz, R. D. S.; Damos, F. S.; dos Santos, A. S.; Franco, D. L.; dos Santos, W. T. P.; J. Braz. Chem. Soc. 2012, 23, 1800.
-
29Felix, F. S.; Angnes, L.; J. Pharm. Sci. 2010, 99, 4784.
-
30Quintino, M. S. M.; Angnes, L.; Electroanalysis 2004, 16, 513.
-
31Ruzicka, J.; Hansen, E. H.; Anal. Chim. Acta 1975, 78, 145.
-
32Wang, J.; Taha, Z.; Anal.Chem. 1991, 63, 1053.
-
33Couto, R. A. S.; Lima, J.; Quinaz, M. B.; Talanta 2016, 146, 801.
-
34Carsol, M. A.; Mascini, M.; Talanta 1998, 47, 335.
-
35Tormin, T. F.; Cunha, R. R.; da Silva, R. A. B.; Munoz, R. A. A.; Richter, E. M.; Sens. Actuators, B 2014, 202, 93.
-
36Richter, E. M.; Tormin, T. F.; Cunha, R. R.; Silva, W. P.; Perez-Junquera, A.; Fanjul-Bolado, P.; Hernandez-Santos, D.; Munoz, R. A. A.; Electroanalysis 2016, 28, 1856.
-
37http://www.dropsens.com/en/accesories_pag.html, acessada em 06/04/2017
» http://www.dropsens.com/en/accesories_pag.html -
38Caramit, R. P.; Lucca, B. G.; Ferreira, V. S.; Munoz, R. A. A.; Richter, E. M.; da Silva, R. A. B.; Electroanalysis 2015, 27, 271.
-
39Dias, A. A.; Cardoso, T. M. G.; Cardoso, R. M.; Duarte, L. C.; Munoz, R. A. A.; Richter, E. M.; Coltro, W. K. T.; Sens. Actuators, B 2016, 226, 196.
-
40Bergamini, M. F.; Santos, A. L.; Stradiotto, N. R.; Zanoni, M. V. B.; J. Pharm. Biomed. Anal. 2007, 43, 315.
-
41Fanjul-Bolado, P.; Lamas-Ardisana, P. J.; Hernandez-Santos, D.; Costa-Garcia, A.; Anal. Chim. Acta 2009, 638, 133.
-
42Stefano, J. S.; Montes, R. H. O.; Richter, E. M.; Munoz, R. A. A.; J. Braz. Chem. Soc. 2014, 25, 484.
-
43Stefano, J. S.; Cordeiro, D. S.; Marra, M. C.; Richter, E. M.; Munoz, R. A. A.; Electroanalysis 2016, 28, 350.
-
44Ke, Q.; Wang, J.; J. Materiomics 2016, 2, 37.
-
45Ferreira, L. M. C.; Felix, F. S.; Angnes, L.; Electroanalysis 2012, 24, 961.
-
46Dos Santos, W. T. P.; Ceolin, M. P.; de Albuquerque, Y. D. T.; Richter, E. M.; Quim. Nova, 2007, 30, 1754.
Datas de Publicação
-
Publicação nesta coleção
Ago 2017
Histórico
-
Recebido
13 Dez 2016 -
Aceito
02 Mar 2017