Abstracts
Nicotine, an oxidizing agent, is certainly one of the most widely used alkaloids in the world. It is, together with its main metabolite, cotinine, responsible for tobacco-dependence. The use of tobacco is closely associated with lung disease, morphological leukocyte modification and generation of oxidant species. The aim of this study was to look for a possible relationship between cotinine, oxidant species generation and oxidative processes. After studying the action of cotinine in some chemical oxidation models and on the enzymatic kinetics of peroxidases (myeloperoxidase and horseradish peroxidase), we concluded that cotinine does not act directly upon H2O2, HOCl, taurine chloramines, horseradish peroxidase or myeloperoxidase.
nicotine; cotinine; myeloperoxidase; ROS; oxidative damage
A nicotina, um agente oxidante, é certamente um dos alcalóides mais amplamente utilizados no mundo. Juntamente com seu principal metabolito, a cotinina, a nicotina é responsável pela tabaco-dependência. O uso de tabaco está intimamente associado a doenças pulmonares, modificações morfológicas em leucócitos e a geração de espécies oxidantes. O principal objetivo deste estudo foi identificar uma possível relação entre cotinina, geração de espécies reativas do oxigênio e processos oxidativos. Após o estudo da ação da cotinina em diferentes modelos químicos e em cinéticas enzimáticas com peroxidases (mieloperoxidase e peroxidase de raiz forte), conclui-se que a cotinina não age diretamente sobre H2O2, HOCl, taurine cloramina, peroxidase de raiz forte ou mieloperoxidase.
nicotina; cotinina; mieloperoxidase; ROS; dano oxidativo
Does cotinine act upon reactive oxygen species and peroxidases?
A cotinina age sobre espécies reativas do oxigênio e peroxidases?
J.C.R. VellosaI, II; N.M. KhalilI, II; L.M. FonsecaII; I.L. BrunettiII; O.M.M.F. OliveiraI, * * Corresponding Author E-mail: olgaenzimologia@yahoo.com.br
IUNESP - Sao Paulo State University, Biochemistry and Technological Chemistry Department, Chemistry Institute, Araraquara-SP, Brazil
IIUNESP - Sao Paulo State University, Clinical Analysis Department, School of Pharmaceutical Sciences, Araraquara-SP, Brazil
ABSTRACT
Nicotine, an oxidizing agent, is certainly one of the most widely used alkaloids in the world. It is, together with its main metabolite, cotinine, responsible for tobacco-dependence. The use of tobacco is closely associated with lung disease, morphological leukocyte modification and generation of oxidant species. The aim of this study was to look for a possible relationship between cotinine, oxidant species generation and oxidative processes. After studying the action of cotinine in some chemical oxidation models and on the enzymatic kinetics of peroxidases (myeloperoxidase and horseradish peroxidase), we concluded that cotinine does not act directly upon H2O2, HOCl, taurine chloramines, horseradish peroxidase or myeloperoxidase.
Keywords: nicotine; cotinine; myeloperoxidase; ROS; oxidative damage.
RESUMO
A nicotina, um agente oxidante, é certamente um dos alcalóides mais amplamente utilizados no mundo. Juntamente com seu principal metabolito, a cotinina, a nicotina é responsável pela tabaco-dependência. O uso de tabaco está intimamente associado a doenças pulmonares, modificações morfológicas em leucócitos e a geração de espécies oxidantes. O principal objetivo deste estudo foi identificar uma possível relação entre cotinina, geração de espécies reativas do oxigênio e processos oxidativos. Após o estudo da ação da cotinina em diferentes modelos químicos e em cinéticas enzimáticas com peroxidases (mieloperoxidase e peroxidase de raiz forte), conclui-se que a cotinina não age diretamente sobre H2O2, HOCl, taurine cloramina, peroxidase de raiz forte ou mieloperoxidase.
Palavras-chave: nicotina; cotinina; mieloperoxidase; ROS; dano oxidativo.
Introduction
Phagocytosis by polymorphonuclear leukocytes is a process that generates reactive oxygen metabolites during the respiratory burst of non-mitochondrial O2 uptake. Simultaneously, an increase in glucose oxidation via the hexose monophosphate shunt, and a decrease in the NADPH/NADP+ ratio, are observed. Two key enzyme systems are involved in these oxidative stress reactions, which produce reactive oxygen species (ROS). First is NADPH oxidase, a mitochondrial membrane multienzyme complex, which catalyses the univalent reduction of molecular oxygen by NADPH and generates O2(sum)-. Subsequently, H2O2, which is also generated during the respiratory burst, is utilized by myeloperoxidase (MPO, an enzyme present in the azurophil granules in neutrophils) in the presence of halide ions such as Cl ?, or Br -, to produce, respectively, HOCl, HOBr, and related oxidative compounds, such as chloramines [1]. During the respiratory burst, a number of enzymatic processes are activated, which then produce oxygenated metabolites. Reactive halogen species (RHS: X2 and HOX, where X represents Cl, Br, or I) are some of the metabolites mediated by neutrophil activation and their respiratory burst [2]. Like other peroxidases (haem proteins that use oxygen as a substrate for oxidative reactions), MPO can also catalyse the oxidation by H2O2 of a number of organic compounds, including phenolic compounds [1].
Despite their physiological role in defence of the organism, reactive oxygen species have been implicated in initiating, accompanying or causing many diseases. There is some evidence that abnormalities in lipid compounds may cause overproduction of ROS and, in turn, antioxidant enzyme activity and lipid peroxidation, and that these phenomena may be related to the pathophysiology of general depression. Free radicals such as the superoxide anion (O2(sum)-) and the hydroxyl radical (HO (sum)), which cause lipid peroxidation, can lead to cell death. Hydrogen peroxide is thought to be the major precursor of highly reactive free radicals, and it has been reported to induce apoptosis in cells of the central nervous system [3].
Oxidative processes play a significant role in atherosclerosis. Oxidized lipids, sterols and proteins have been detected at high levels in human lesions. The interaction of the extra-cellular matrix (ECM) with cells regulates their adhesion, migration and proliferation, and it is believed that damage to vascular matrix components is a factor in the development of atherosclerosis. There is some evidence that the heme enzyme MPO, released by activated monocytes (and possibly macrophages) has a role in oxidative events within the artery wall. As MPO is released extracellularly, and is highly basic, it might be expected to associate with poly-anionic matrix components thereby localizing damage to the ECM [4].
MPO has recently attracted considerable attention as a possible mediator of coronary artery disease and acute coronary syndromes. Active MPO, hypochlorite-modified proteins and peptides, and chlorinated oxidized lipid species with proatherogenic properties have been detected in human atherosclerotic lesions [5].Cigarette smoking is a risk factor for the development of atherosclerosis. Possible mechanisms for this include leukocyte and platelet activation and/or damage to the endothelium, any of which may contribute to thrombosis and changes in haemostasis [6].
Nicotine [3 - (1- methyl 2 - pyrrolidinyl) pyridine] is the principal alkaloid in tobacco and is present as a major component of tobacco smoke. It is absorbed in measurable quantities by both active and passive smokers, the latter having been shown to inhale an amount of nicotine proportional to the product of concentration, duration of exposure and respiration rate [7]. Recently, it has been shown in human liver microsomes that nicotine and cotinine [1 methyl 5 - (3 - pyridinyl)- 2 - pyrrolidinone] are oxidized primarily via CYP 2A6 [8]. As shown in Scheme 1, nicotine is primarily metabolized to cotinine, and cotinine is further metabolized to trans-3'-hydroxycotinine in the human liver, which is a major metabolite of nicotine in humans [9]. Cotinine is used as a marker of nicotine exposure, both in smokers and in nonsmokers, as it is one of the main metabolites of nicotine and has a longer half-life than its precursor [10].
Nicotine administration may result in oxidative stress by inducing the generation of reactive oxygen species in the peripheral and central nervous system [11]. Cotinine has been reported to have pharmacological activity. It interferes with the release of brain neurotransmitters and affects enzymes involved in the synthesis of estrogen and testosterone, decreases vascular resistance and blood pressure in animals, and has been reported to have effects on cognitive performance and to modify nicotine withdrawal symptoms in abstinent smokers [10].
The major inducer of oral cancer is exposure to tobacco, which is responsible for 50 to 90% of cases worldwide [12]. Cigarette smoking results in an increased cumulative exposure to ROS and, because its accumulation, it would seem cigarette smokers may have an increased requirement for antioxidant nutrients [13].
Owing to the involvement of cigarette smoking in oxidative damage, it was seen as highly relevant to determine whether cotinine, one of the main metabolites of nicotine, has any effect on ROS activity.
Experimental details
Apparatus and Materials
Both enzymes and chemicals were purchased from SIGMA CHEMICALS CO.
Spectrophotometric assays were performed in a diode array spectrophotometer (HP 8453), monitored by a PC Pentium III from Hewlett Packard. Chemiluminescence assays were performed in a 1251 Luminometer from BioOrbit, monitored by a workstation with the program Multiuse v 2.0.
Hydrogen peroxide scavenging assay
TNB (5-thio-2-nitrobenzoic acid), was prepared by incubating, 1 mmol.L-1 DTNB (5,5'-dithio-2-nitrobenzoic acid), 20 mmol.L-1 NaBH4 in 50 mmol.L-1 potassium phosphate solution, pH 6.6, with 5 mmol.L-1 EDTA (ethylene diamine tetraacetic acid), at 370C for 30 min. TNB oxidation is widely used to verify a possible hypochlorous acid-scavenging activity [14]. TNB is oxidized by H2O2 too, resulting in a decreasing absorbance at 412 nm, because the product DTNB does not absorb at this wavelength. We tested the effect of adding cotinine at 0.1 mmol.L-1. TNB concentration was measured by its molar absorption coefficient (13600 mol-1L.cm-1) at 412 nm [14].
Hypochlorous acid and chloramine scavenging assays
Potassium iodide (KI) is oxidized by HOCl or chloramine (taurine chloramine-TauCl), decreasing the absorbance at 350 nm. Chloramine was produced by incubation of 0.05 mmol.L-1 HOCl and 0.2 mmol.L-1 taurine (Tau) for 1 min. Both reactants were prepared in PBS-D (Dulbecco's phosphate-buffered saline) [15].
Chemiluminescence from HRP kinetics
The solutions (luminol, horseradish peroxidase - HRP and hydrogen peroxide - H2O2) were prepared in water and their molar concentrations were determined by the molar absorption coefficients:
Luminol: e 347 nm = 7636 mol-1.L.cm-1 [16]
H2O2: e 230 nm = 80 mol-1.L.cm-1 [17]
HRP : e 403 nm = 1,02 x 105 mol-1.L.cm-1 [18]
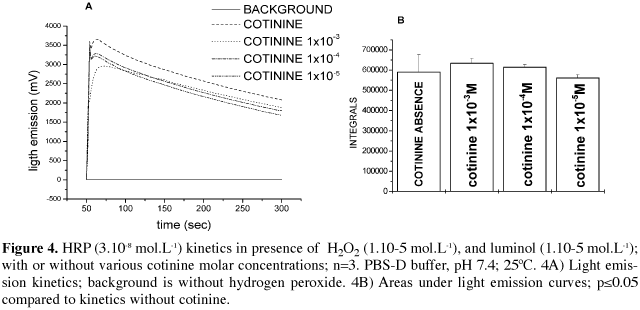
HRP is an important model for peroxidase studies, so we carried out some kinetic studies with this classic working enzyme. HRP/H2O2/luminol chemiluminescence [19] studies were performed in the presence (1 mmol.L-1, 0.1 mmol.L-1 and 0.01 mmol.L-1) and absence of cotinine, in PBS-D Buffer, pH 7.4, at 250C. Light intensity was registered as mV and integrated by Multiuse v 2.0.
Spectrophotometric HRP kinetics
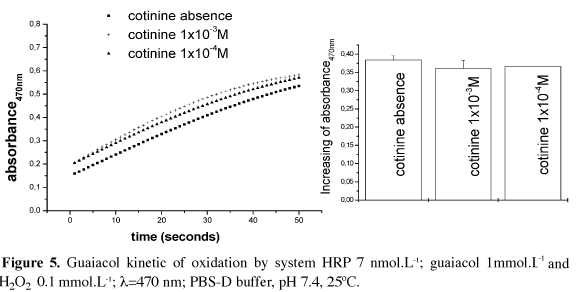
HRP and hydrogen peroxide solutions were prepared as described before. Guaiacol was diluted in 50 mmol.L-1 potassium phosphate buffer 50, pH 7.0. Reactions were performed in PBS-D, pH 7,4, 250C, monitored spectrophotometrically at 470 nm. The absorbance increased due to guaiacol oxidation by HRP and H2O2.
MPO kinetics
MPO activity was determined by the guaiacol assay as recommended by Sigma Chemical Co. Luminol, and H2O2 solutions were prepared as described before.
MPO/H2O2/luminol chemiluminescence [19] studies were performed in the presence (0.1 mmol.L-1) and absence of cotinine, in PBS-D Buffer, pH 7.4, at 250C. Light intensity was registered as mV and kinetics integrals were analyzed by the program Multiuse v 2.0.
Results and Discussion
The results are all expressed as means ± SD (standart deviation). Data were analyzed with the graphical and statistical modeling package "ORIGIN5.0", and differences were tested by ANOVA (analysis of variance), significance being declosed at p< 0.05.
To enable measurement of concentrations of cotinine solutions, we determined its molar absorption coefficient in water. From the slope of trace linear plots of the absorbance at 261 nm versus the molar concentration of cotinine, we obtained the result 2196.3 + 10.5 mol-1.L.cm-1.
The H2O2 reactions were followed by the decrease in absorbance of TNB at 412 nm, as hydrogen peroxide oxidized TNB to DTNB; as can be seen in Figure 1, there was no significative difference in the absorbance at any wavelength, with or without cotinine.
The HOCl or TauCl reactions were followed by decreasing absorbance of KI at 350 nm, as HOCl or TauCl oxidized KI. As observed in Figures 2 and 3, cotinine had no effect on the HOCl and TauCl oxidative capacities.
HRP kinetics were monitored by chemiluminescence, comparing the light intensity (mV) in the presence and absence of cotinine at various molar concentrations. As shown figure 4, cotinine was not able to stimulate or inhibit luminol oxidation by HRP.
The HRP reaction was also studied spectrophotometrically by guaiacol oxidation, which generates a product with an absorbance at 470 nm. The absorbance changes in the presence and absence of cotinine at various molar concentrations were compared, (Figure 5) and it is clear, as in chemiluminescent assay, that cotinine did not affect HRP activity.
MPO kinetics was also monitored by chemiluminescence, light intensity (mV) being compared in the presence and absence of cotinine. Myeloperoxidase is an important component of ROS generation in living organisms, which is able to generate hypochlorous acid, a strong deleterious ROS. So it is important to detect different substances in tobacco that are able to increase the oxidative process. In this assay, shown in Figure 6, it is clear that cotinine, a nicotine-derived product, does not act upon luminol oxidation by MPO.
Conclusion
Tobacco smoking is one of most widespread habits in the world. It ocurrs because it is an easily acquired addiction, being, like alcohol, a permitted drug. It has consequences for public health, representing heavy cost that the government has to cover. Tobacco has many potentially oxidants and carcinogenic substances and it is necessary to know how these substances affect free radical generation in the human body. Cotinine was in others enzymes systems (like cytochrome P450) but not on ours studied systems, then, this study is relevant so far as: i) it may help elucidate this involvement in oxidative generation and its consequent damage; ii) it determined the molar abortion coefficient of cotinine in water, 2196.3 + 10.5 mol-1.L.cm-1.
Cotinine, as the main product of nicotine metabolism, could be involved in the oxidative burst, making it necessary to study its influence on ROS action and generation. We have carried out chemical experiments to elucidate the interference of cotinine in the oxidative activity of three major oxidative species that participate on myeloperoxidase ROS generation: hydrogen peroxide, hypochlorous acid and taurine chloramine. The experimental results show that cotinine has no influence on oxidative action of these molecules. We can conclude, too, from these results that cotinine neither inhibits nor stimulates peroxidase (HRP and MPO) activity. Further work will be needed to confirm that cotinine is not one of the molecules involved in oxidative stress promoted by tobacco smoking.
Acknowledgement
J. C. R. Vellosa is grateful to CNPq/PIBIC for his scholarship.
Recebido em: 15/12/2006
Aceito em:15/03/2007
- [1] C. Capeilleare-Blandin, Biochem. J. 336 (1998) 395.
- [2] Y.R. Chen, L.J. Deterding, B.E. Sturgeon, K.B. Tomer, R.P. Mason, J. Biol. Chem. 277 (2002) 29781.
- [3] J. Benedi´, R. Arroyo, C. Romero, S.M. Aragon, A.M. Villar, Life Sci. 75 (2004) 1263.
- [4] A.A. Woods, S.M. Linton, M.J. Davies, Biochem. J. 370 (2003) 729.
- [5] A.K Thukkani, J. Mchowat, F.F. Hsu, M.L. Brennan, S.L. Hazen, D.A. Ford, Circulation 108 (2003) 3128.
- [6] A.D. Blann, U. Kirkpatrick, C. Devine, S. Naser, C.N. Mccollum, Atherosclerosis 141 (1998) 133.
- [7] M.C Bentley, M. Abrar, M. Kelk, J. Cook, K. Phillips, J. Chromat. B 723 (1999) 185.
- [8] E.S. Messina, R.F. Tyndale, E.M. Sellers, Pharmacol. Expther. 282 (1997) 1608.
- [9] M. Nakajima, T. Yamamoto, K. Nunoya, T. Yokoi, K. Nagashima, K. Inoue, Y. Funae, N. Shimada, T. Kamataki, Y. Kuroiwa, J. Pharmacol. Exp. Ther. 277 (1996) 1010.
- [10] S. Zevin, P. Jacob III, P. Geppetti, N.L. Benowitz, Drug And Alcohol Dependence 60 (2000) 13.
- [11] M.B. Newman, G.W. Arendash, R.D. Shytle, P.C. Bickford, T. Tighe, P.R. Sanberg, Life Sci. 71 (2002) 2807.
- [12] A.Z. Reznick, I. Klein, J.P. Eiserich, C.E. Cross, R.M. Nagler, Free Radic. Biol. Med. 34 (2003) 377.
- [13] G. Kelly, Altern. Med. Rev. 8 (2003) 43.
- [14] T. Ching, J. Jong, A. Bast, Anal. Biochem. 218 (1994) 377.
- [15] M.B. Babior, H.J. Cohen, Methods In Hematology: Leukocyte Function 13 (1981).
- [16] R.C. Allen, L.D. Loose, Biochem. Biophys. Res. Commun. 69 (1976) 245.
- [17] E.P. Brestel, Biochem. Biophys. Res. Comm. 126 (1985) 482.
- [18] P.I. Ohlson, K.G. Paul, Acta Chem. Scand. Ser. B 31(1976) 373.
- [19] I.L. Brunetti, O.M.M.F. Oliveira, Rev. Cienc. Farm. 16 (1996) 55.
Publication Dates
-
Publication in this collection
04 June 2007 -
Date of issue
2007
History
-
Accepted
15 Mar 2007 -
Received
15 Dec 2006