Resumos
A displasia camptomélica pertence a um grupo heterogêneo e raro de displasias esqueléticas letais, que se caracterizam pelo desenvolvimento anormal dos ossos e das cartilagens. É causada por uma mutação no gene Sox9 (SRY-like HMG [high-mobility group] BOX 9) do cromossomo 17 e transmitida pela via autossômica dominante. Apresenta como principais características o encurtamento e o encurvamento dos ossos longos, principalmente nos membros inferiores. Também está associada a outras graves malformações esqueléticas e extra-esqueléticas. O estudo do cariótipo pode revelar incompatibilidade entre o genótipo e o fenótipo genital. A maioria dos portadores morre nos períodos fetal e neonatal precoce. A ultra-sonografia é essencial para a elucidação diagnóstica pré-natal.
Osteocondrodisplasias; Osteocondrodisplasias; Doenças do desenvolvimento ósseo; Anormalidades congênitas; Anormalidades múltiplas; Ultra-sonografia pré-natal
Camptomelic dysplasia belongs to a heterogeneous and rare group of lethal skeletal dysplasias, characterized by abnormal development of bones and cartilages. It is caused by a mutation in gene Sox9 (SRY-like HMG [high-mobility group] BOX 9) of chromosome 17 and it is transmitted as an autosomal dominant trait. Its main characteristics are the shortening and bowing of the long bones, principally the lower limbs. It is also associated with other severe skeletal and extraskeletal malformations. Karyotype study may reveal sex reversal. The majority of carriers die during the fetal and early neonatal periods. Ultrasound is essential to elucidate a prenatal diagnosis.
Osteochondrodysplasias; Osteochondrodysplasias; Bone diseases, developmental; Congenital abnormalities; Abnormalities, multiple; Ultrasonography, prenatal
RELATO DE CASO
Diagnóstico pré-natal de displasia camptomélica: relato de caso
Prenatal diagnosis of camptomelic dysplasia: a case report
Tadeu CoutinhoI; Conrado Milani CoutinhoII; Larissa Milani CoutinhoIII
IProfessor Associado, Chefe do Serviço e Coordenador da Residência de Obstetrícia da Faculdade de Medicina da Universidade Federal de Juiz de Fora UFJF Juiz de Fora (MG), Brasil
IIMédico Assistente do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo USP Ribeirão Preto (SP), Brasil
IIIAcadêmica de Medicina e Monitora da Disciplina de Obstetrícia da Faculdade de Medicina da Universidade Federal de Juiz de Fora UFJF Juiz de Fora (MG), Brasil
Correspondência Correspondência: Tadeu Coutinho Rua Batista de Oliveira, 1.070/1.502 Granbery CEP: 36010-532 Juiz de Fora/MG Fone: (32) 3211-9552 E-mail: coutinho@nextwave.com.br
RESUMO
A displasia camptomélica pertence a um grupo heterogêneo e raro de displasias esqueléticas letais, que se caracterizam pelo desenvolvimento anormal dos ossos e das cartilagens. É causada por uma mutação no gene Sox9 (SRY-like HMG [high-mobility group] BOX 9) do cromossomo 17 e transmitida pela via autossômica dominante. Apresenta como principais características o encurtamento e o encurvamento dos ossos longos, principalmente nos membros inferiores. Também está associada a outras graves malformações esqueléticas e extra-esqueléticas. O estudo do cariótipo pode revelar incompatibilidade entre o genótipo e o fenótipo genital. A maioria dos portadores morre nos períodos fetal e neonatal precoce. A ultra-sonografia é essencial para a elucidação diagnóstica pré-natal.
Palavras-chave: Osteocondrodisplasias/diagnóstico, Osteocondrodisplasias/ultra-sonografia, Doenças do desenvolvimento ósseo, Anormalidades congênitas, Anormalidades múltiplas, Ultra-sonografia pré-natal
ABSTRACT
Camptomelic dysplasia belongs to a heterogeneous and rare group of lethal skeletal dysplasias, characterized by abnormal development of bones and cartilages. It is caused by a mutation in gene Sox9 (SRY-like HMG [high-mobility group] BOX 9) of chromosome 17 and it is transmitted as an autosomal dominant trait. Its main characteristics are the shortening and bowing of the long bones, principally the lower limbs. It is also associated with other severe skeletal and extraskeletal malformations. Karyotype study may reveal sex reversal. The majority of carriers die during the fetal and early neonatal periods. Ultrasound is essential to elucidate a prenatal diagnosis.
Keywords: Osteochondrodysplasias/diagnosis, Osteochondrodysplasias/ultrasonography, Bone diseases, developmental, Congenital abnormalities, Abnormalities, multiple, Ultrasonography, prenatal
Introdução
A displasia camptomélica ou campomélica é uma osteocondrodisplasia muito rara e freqüentemente letal, caracterizada pelo desenvolvimento de curvatura anormal dos ossos longos fetais, particularmente do fêmur e da tíbia. Devido à presença de uma grande variedade de malformações esqueléticas e extra-esqueléticas graves, alguns autores preferem utilizar a denominação síndrome camptomélica. Foi descrita inicialmente na década de 1970 e a denominação deriva das palavras gregas kamptós (curvo) e mélos (membro)1. A sua incidência varia, na literatura, entre 1/10.000 nascidos vivos e 2/1.000.000 nascidos vivos2,3.
Ao lado das alterações esqueléticas predominantes (como encurvamento e diminuição do comprimento de ossos longos, hipoplasia das escápulas, anomalias dos ossos da pelve e mineralização deficiente dos pedículos torácicos), anomalias faciais, cardíacas, nervosas, respiratórias e genitourinárias estão freqüentemente associadas à displasia camptomélica. São também comuns o desenvolvimento de incompatibilidade entre o genótipo e o fenótipo genital, com cariótipo 46XY e genitália feminina, e o óbito no período neonatal, devido à insuficiência respiratória4,5. Embora a literatura especializada mostre que a definição diagnóstica da maioria dos casos de displasia camptomélica ocorre só após o nascimento6, a ampla variedade de anomalias e os altos índices de mortalidades fetal e neonatal requerem um diagnóstico antenatal precoce. Com essa finalidade, a utilização cada vez mais precoce da ultra-sonografia obstétrica exerce um papel fundamental na suspeita inicial e nos diagnósticos definitivo e diferencial da displasia camptomélica, como exposto no presente relato de caso.
Relato de caso
Paciente com 34 anos de idade, secundigesta, com história de parto cesáreo a termo e recém-nascido normal, compareceu à primeira consulta pré-natal na sexta semana gestacional. A anamnese revelou ausência de consangüinidade do casal e de malformações fetais, tanto na família materna quanto na paterna. As propedêuticas clínica e complementar empreendidas nas duas primeiras consultas da assistência pré-natal permitiram avaliar a gestação, inicialmente, como de baixo risco.
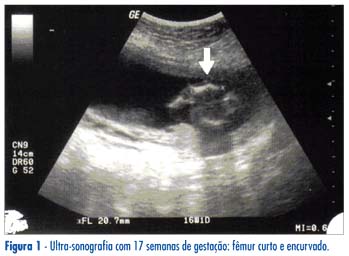
Um exame ultra-sonográfico rotineiro realizado na 11ª semana de gestação confirmou a idade gestacional e revelou um aumento da translucência nucal (TN, 3,7 mm). Foi indicada e realizada a biópsia de vilo corial (BVC). Durante a BVC, a avaliação ecográfica ratificou a alteração da TN (4,8 mm) e demonstrou uma diminuição do comprimento femoral. A análise do cariótipo fetal foi normal e revelou sexo genético fetal masculino (46XY). Uma ultra-sonografia morfológica realizada mais precocemente (17ª semana) comprovou o encurtamento femoral bilateral (Figura 1: 20 mm; 5º percentil=21 mm) e reafirmou a existência das deformidades nos eixos dos ossos longos dos membros inferiores, além de revelar a ausência da genitália masculina externa.
Frente à confirmação desses achados em exames ecográficos posteriores, a família foi cientificada do provável diagnóstico de displasia camptomélica. Após a certificação da inexistência de aumento de risco para a saúde materna, a família foi informada acerca das múltiplas anomalias e da letalidade associadas à doença. A realização de uma ecocardiografia fetal excluiu malformações cardíacas congênitas. A partir da 33ª semana, foi diagnosticada a polidramnia, que se manteve estável até o termo.
A paciente foi submetida à cesariana na 39ª semana de gestação. O recém-nascido apresentou peso de 2.885 g, comprimento de 39 cm e índices de Apgar de 4 e 7 no primeiro e no quinto minuto pós-parto, respectivamente.
O exame físico pós-parto confirmou as anormalidades dos membros inferiores e a genitália externa feminina, além de revelar macrocefalia, fenda palatina posterior, implantação baixa das orelhas, tórax em sino e hipoplasia das escápulas (Figuras 2 e 3). Devido ao desconforto respiratório, recebeu assistência respiratória imediata, com entubação orotraqueal na unidade de terapia intensiva neonatal.
As radiografias realizadas no neonato revelaram anormalidades em crânio (desproporção crânio-facial) e em membros inferiores (deformidades de fêmures e tíbias, com hipoplasia e encurtamento das fíbulas). Foram normais os resultados da ecocardiografia e da ultra-sonografia abdominal e transfontanela, excluindo a existência de hidrocefalia. No oitavo dia pós-parto, a tomografia computadorizada revelou hemorragia ventricular cerebral e uma segunda radiografia de tórax diagnosticou atelectasia total do pulmão direito. Devido ao agravamento paulatino do quadro de insuficiência respiratória, o óbito ocorreu no 12º dia pós-nascimento.
A paciente assinou termo de compromisso livre e esclarecido autorizando a publicação do relato de caso.
Discussão
As displasias ósseas incluem um conjunto amplo e heterogêneo de modificações congênitas do metabolismo do osso e da cartilagem. Dentre elas, a displasia camptomélica é uma rara alteração que, devido à sua associação com altos índices das mortalidades fetal e neonatal, requer um diagnóstico antenatal precoce. Ademais, a gestação e o nascimento de um portador da síndrome e, também, a expectativa sobre a possibilidade da sua ocorrência em gestações futuras geram uma experiência familiar impactante. A recorrência da displasia camptomélica em irmãos é de 5%7.
As mortes neonatais precoces ocorrem em decorrência de uma grave insuficiência respiratória. As principais causas anatômicas para esses óbitos são as dimensões reduzidas da caixa torácica, a hipoplasia pulmonar e a laringotraqueomalacia, com estreitamento das vias aéreas1. Essas alterações se originam do desenvolvimento anormal da cartilagem laringotraqueobrônquica e podem resultar em apnéia, atelectasia, aspiração e pneumonia2.
Além das anomalias mais proeminentes do esqueleto apendicular que caracterizam a displasia camptomélica, uma série de outras alterações pode ser atribuída à síndrome, envolvendo uma gama de defeitos esqueléticos e não esqueléticos.
As formações cartilaginosa e óssea incompletas podem resultar em tórax pequeno e em forma de sino, escápula hipoplásica, cifoescoliose, adelgaçamento e redução do número das costelas (11 pares), pés tortos eqüinovaros, hipoplasias dos ossos da pelve, entre outras alterações. Há relatos de complicações neurocirúrgicas, devido à ossificação incompleta dos pedículos vertebrais cervicais, que resultam em instabilidade cervical e cifose congênitas8. São também observados: macrocefalia, hipertelorismo, ponte nasal achatada, micrognatia, fenda palatina, orelhas de implantação baixa, hidrocefalia, ausência do bulbo olfatório, poligiria. Em um terço dos portadores de displasia camptomélica, foram notadas malformações cardíacas, como defeitos septais, tetralogia de Fallot e persistência do ducto arterial. O desenvolvimento da incompatibilidade entre o genótipo e o fenótipo genital, por sua vez, pode ser demonstrado pela comparação entre os achados ultra-sonográficos e o estudo do cariótipo. Entre 65 e 75% dos portadores do cariótipo 46XY possuem fenótipo feminino ou ambigüidade genital9-13. A polidramnia tem sido reportada entre 25 e 48% dos casos4.
O mecanismo de herança da displasia camptomélica foi motivo de controvérsia durante muito tempo14. Apesar de alguns estudos terem demonstrado a existência de pares de irmãos com displasia camptomélica, sugerindo que esta síndrome poderia ser adquirida de modo recessivo15,16, pesquisas posteriores indicaram tratar-se de uma condição autossômica dominante esporádica4,7. Os casos severos de displasia camptomélica são herdados pela via autossômica dominante, por meio da mutação do gene Sox9 (SRY-like HMG [high-mobility group] BOX 9), localizado no braço longo do cromossomo 17, e resultam em óbito nos primeiros dias de vida5. As formas menos graves da doença adviriam de uma translocação balanceada t (13;17), com uma sobrevida de até três décadas5.
O gene SRY (sex-determining region of Y chromosome), também denominado TDF (testis-determining factor), é implicado na formação dos testículos17. As mutações no cromossomo 17 levam à inativação do gene Sox9, mesmo quando ocorrem em apenas um dos alelos, podendo-se concluir pela existência de uma haploinsuficiência do gene em questão10,14,18. Como o gene Sox9 atua tanto na formação óssea como no desenvolvimento testicular, essas mutações são responsáveis pelas várias anomalias do esqueleto e dos órgãos sexuais que estão associadas à displasia camptomélica. A conseqüente anormalidade dos testículos fetais altera a produção dos hormônios masculinos e resulta no desenvolvimento feminino incluindo ovários, trompas, útero e vagina de um indivíduo genotipicamente XY19-21.
Muitas das malformações fetais graves podem ser diagnosticadas ou, ao menos, suspeitadas durante o exame ultra-sonográfico de alta resolução entre 11 e 14 semanas de gestação22. A partir da 11ª semana de gravidez, os ossos longos podem ser medidos com acurácia satisfatória23. A análise de todas as extremidades confirmará a presença de displasia esquelética e contribuirá para o diagnóstico diferencial, mas a realização de exames seriados pode ser necessária24,25. O diagnóstico diferencial antenatal inclui a displasia torácica asfixiante, a hipofosfatasia, a osteogênese imperfeita tipos 1 e 2 e variedades não classificadas de encurvamento de ossos longos6.
Devido à presença de um quadro clínico bastante característico, a ecografia exerce papel fundamental na suspeita inicial e nos diagnósticos definitivo e diferencial da displasia camptomélica.
No caso aqui descrito, o achado ultra-sonográfico inicial foi um aumento da TN, que é uma expressão fenotípica comum da trissomia do cromossomo 21 e de outras cromossomopatias, mas também está associado a síndromes genéticas e a malformações fetais graves. A prevalência de malformações e o mau prognóstico da gestação crescem exponencialmente com o aumento da espessura da TN. A anormalidade da TN nos casos de displasias esqueléticas pode ser explicada pela congestão venosa na cabeça e na região cervical que é resultante da constrição torácica fetal. Porém, uma alteração da matriz celular associada a modificações no metabolismo do colágeno pode também participar da fisiopatologia do aumento da TN nessas patologias. Após a constatação de um aumento da TN, a avaliação do cariótipo fetal deve ser oferecida à família.
Neste relato, a evidência ecográfica de encurtamento femoral reforçou a indicação de estudo do cariótipo fetal, que foi realizado pela BVC, e, também, de uma pesquisa mais acurada de alterações esqueléticas que pudessem ser produzidas pelas diferentes formas de displasias esqueléticas.
Na maioria dos casos de displasia camptomélica descritos na literatura, a definição do diagnóstico só ocorreu após o nascimento8. Entretanto, por ser uma condição grave, que abrange uma ampla variedade de anomalias e um elevado obituário neonatal, é fundamental a confirmação diagnóstica da displasia camptomélica durante o período pré-natal.
O caráter essencial da ultra-sonografia na pesquisa diagnóstica antenatal da síndrome reforça a necessidade da busca cotidiana pela melhoria da qualidade dos exames ecográficos que são realizados rotineiramente durante a assistência pré-natal. O advento da ecografia tridimensional possibilitou avaliar, de forma mais acurada, o volume de diversos órgãos fetais, permitindo um diagnóstico mais precoce e preciso dos distúrbios do crescimento e desenvolvimento25. O conhecimento adequado da morfologia e do desenvolvimento normais do feto é também imprescindível para evitar potenciais erros diagnósticos. A presença de achados ultra-sonográficos sugestivos da síndrome indica a realização da cariotipagem fetal para a determinação do diagnóstico definitivo e do prognóstico do concepto.
O diagnóstico anteparto resulta na precocidade do aconselhamento parental e no planejamento da conduta obstétrica posterior, além dos cuidados pós-natais. Permite também o aconselhamento adequado sobre o futuro reprodutivo do casal.
Recebido: 12/11/07
Aceito com modificações: 16/6/08
- 1. Maroteuax P, Spranger JW, Opitz JM, Kucera J, Lowry RB, Schimke R, et al. Le syndrome campomélique. Presse Méd. 1971;79(25):1157-62.
- 2. Eger KJ. Campomelic dysplasia. J Diagn Med Sonogr. 2005;21(4):343-9.
- 3. Natasha G, Ghai R, Shah D, Kiran PS. Camptomelic dysplasia: prenatal diagnosis by ultrasound. Skeletal Radiol. 2006;35(9):699-701.
- 4. Mansour S, Hall CM, Pembrey ME, Young ID. A clinical and genetic study of campomelic dysplasia. J Med Genet. 1995;32(6):415-20.
- 5. Kos R, Medjo B, Grković S, Nikolić D, Sajić S, Ilić J. Camptomelic dysplasia: a case report. Srp Arh Celok Lek. 2007;135(5-6):335-8.
- 6. Nagar AM, Sangle PM, Morani AC, Rajpal ND. Antenatal diagnosis of camptomelic dysplasia. J Postgrad Med. 2006;52(1): 69-70.
- 7. Lynch SA, Gaunt ML, Minford AM. Campomelic dysplasia: evidence of autosomal dominant inheritance. J Med Genet. 1993;30(8):683-6.
- 8. Lekovic GP, Mariwalla NR, Horn EM, Chang S, Rekate HL, Theodore N. Skeletal dysplasia involving the subaxial cervical spine. Report of two cases and review of the literature. Neurosurg Focus. 2006;20(2):E8.
- 9. Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79(6):1111-20.
- 10. Iravani S, Debich-Spicer D, Gilbert-Barness E. Pathological case of the month. Campomelic dysplasia. Arch Pediatr Adolesc Med. 2000;154(7):747-8.
- 11. Velagaleti GV, Bien-Willner GA, Northup JK, Lockhart LH, Hawkins JC, Jalal SM, et al. Position effects due to chromosome breakpoints that map approximately 900 Kb upstream and approximately 1.3 Mb downstream of SOX9 in two patients with campomelic dysplasia. Am J Hum Genet. 2005;76(4):652-62.
- 12. Banu NA, Khatoon S, Qadir E, Rahman MM, Khan MA, Islam MZ, et al. Camptomelic dysplasia with sex reversal. Mymensingh Med J. 2005;14(1):80-3.
- 13. Lipay MVN, Bianco B, Verreschi ITN. Disgenesias gonadais e tumores: aspectos genéticos e clínicos. Arq Bras Endocrinol Metab. 2005;49(1):60-70.
- 14. Shafai T. Letter: Camptomelic syndrome in siblings. J Pediatr. 1976;89(3):512-3.
- 15. Mellows HJ, Pryse-Davies J, Bennet MJ, Carter CO. The camptomelic syndrome in two female siblings. Clin Genet. 1980;18(2):137-41.
- 16. Cunningham FG, Leveno KJ, Bloom SL, Hauth JC, Gilstrap LC 3rd, Wenstrom KD. Williams obstetrics. 22nd ed. Boston: McGraw-Hill; 2005.
- 17. Foster JW. Mutations in SOX9 cause both autosomal sex reversal and campomelic dysplasia. Acta Paediatr Jpn. 1996:38(4):405-11.
- 18. Pfeifer D, Kist R, Dewar K, Devon K, Lander ES, Birren B, et al. Campomelic dysplasia translocation breakpoints are scattered over 1 Mb proximal to SOX9: evidence for an extended control region. Am J Hum Genet. 1999;65(1):111-24.
- 19. Mansour S, Offiah AC, McDowall S, Sim P, Tolmie J, Hall C. The phenotype of survivors of campomelic dysplasia. J Med Genet. 2002;39(8):597-602.
- 20. Ninomiya S, Yokoyama Y, Teraoka M, Mori R, Inoue C, Yamashita S, et al. A novel mutation (296 del G) of the SOX9 gene in a patient with campomelic syndrome and sex reversal. Clin Genet. 2000;58(3):224-7.
- 21. Fong KW, Toi A, Salem S, Hornberger LK, Chitayat D, Keating SJ, et al. Detection of fetal structural abnormalities with US during early pregnancy. Radiographics. 2004;24(1):157-74.
- 22. Zorzoli A, Kustermann A, Caravelli E, Corso FE, Fogliani R, Aimi G, et al. Measurements of fetal limb bones in early pregnancy. Ultrasound Obstet Gynecol. 1994;4(1):29-33.
- 23. Spirt BA, Oliphant M, Gottlieb RH, Gordon LP. Prenatal sonographic evaluation of short-limbed dwarfism: an algorithmic approach. Radiographics. 1990;10(2):217-36.
- 24. Bowerman RA. Anomalies of the fetal skeleton: sonographic findings. AJR Am J Roentgenol. 1995;164(4):973-9.
- 25. Seow KM, Huang LW, Lin YH, Pan HS, Tsai YL, Hwang JL. Prenatal three-dimensional ultrasound diagnosis of a camptomelic dysplasia. Arch Gynecol Obstet. 2004;269(2):142-4.
Datas de Publicação
-
Publicação nesta coleção
15 Ago 2008 -
Data do Fascículo
Maio 2008
Histórico
-
Aceito
16 Jun 2008 -
Recebido
12 Nov 2007