Abstract
Several human genetic syndromes have long been recognized to be defective in DNA repair mechanisms. This was first discovered by Cleaver (1968), who showed that cells from patients with xeroderma pigmentosum (XP) were defective for the ability to remove ultraviolet (UV)-induced lesions from their genome. Since then, new discoveries have promoted DNA repair studies to one of the most exciting areas of molecular biology. The present work intends to give a brief summary of the main known human genetic diseases related to DNA repair and how they may be linked to acquired diseases such as cancer
MINI-REVIEW
Human DNA repair diseases: From genome instability to cancer
Carlos R. Machado1 and Carlos F.M. Menck 2
1 Laboratório de Genética-Bioquímica, Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brasil.
2 Departamento de Microbiologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, Av. Prof. Lineu Prestes, 1374, 05508-900 São Paulo, SP, Brasil. Tel.: 55.11.818.7499. Fax: 55.11.818.7354. E.mail: cfmmenck@usp.br.
Several human genetic syndromes have long been recognized to be defective in DNA repair mechanisms. This was first discovered by Cleaver (1968), who showed that cells from patients with xeroderma pigmentosum (XP) were defective for the ability to remove ultraviolet (UV)-induced lesions from their genome. Since then, new discoveries have promoted DNA repair studies to one of the most exciting areas of molecular biology. The present work intends to give a brief summary of the main known human genetic diseases related to DNA repair and how they may be linked to acquired diseases such as cancer.
Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy
Lesions that cause gross distortions on the DNA double helix, such as pyrimidine dimers induced by UV irradiation, are recognized and excised from DNA by a complex mechanism known as nucleotide excision repair. Three human genetic disorders are associated with defects on nucleotide excision repair: xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD). For a summary see Table I.
*These genes are components of the transcription factor TFIIH.
XP individuals are highly sensitive, presenting dramatic pigmentary skin disturbances, specially at regions exposed to sunlight. There is a great increase in the incidence of both benign and malignant skin tumors and frequently these clinical features are accompanied by neurological abnormalities and premature aging. Fibroblasts from these patients are sensitive to UV light and have reduced levels of nucleotide excision repair. They also show an increased frequency of mutagenesis after UV, which correlates well with the patient clinical feature of increased skin carcinogenesis. The XP syndrome has an autosomal recessive inheritance and seven distinct groups of genetic complementation groups were identified, named XPA to XPG. Few patients, known as XP variants (XPV), however, have normal nucleotide excision repair levels and may have a defect on the ability to replicate damaged DNA (Bootsma and Hoeijmakers, 1991).
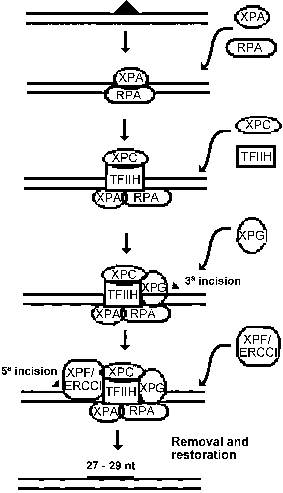
The genes related to XP have recently been cloned, except for XPE and XPV, and have revealed strong homologies with DNA repair genes from other eukaryotic organisms. The concerted action of the XP proteins during nucleotide excision repair is presented as a hypothetical model in Figure 1. The XPA protein in association with RPA protein is responsible for the binding to the damaged DNA (Li et al., 1995). The XPA-RPA complex also has interactions with TFIIH (Park et al., 1995), XPF-ERCC1 (Park and Sancar, 1994) and XPG (He et al., 1995), that play different roles on DNA repair, as described below. Thus, the complex might constitute the nucleation component for the remaining subunits of excision repair.
The fact that XPB and XPD proteins are subunits of the RNA polymerase II transcription factor TFIIH is in agreement with the discovery of the preferential nucleotide excision repair of transcriptionally active genes made by Bohr and his colleagues in 1985. This kind of repair has been denominated "transcription coupled repair" in order to differentiate from the repair of the rest of the genome, known as overall or general repair. The most likely function of TFIIH in transcription is promoter clearance, which is the reaction encompassing the phosphorylation of C-terminal domain of RNA polymerase II, the disruption of the initiation complex and the synthesis of a transcript 30-50 nucleotides in length (Goodrich and Tjian, 1994). For the excision repair, TFIIH participates in the formation of the pre-incision complex after its recruitment by XPA protein to the damaged site. It seems that the helicase activities of XPB (3-5 direction) and XPD (5-3) may be important functions necessary for the role of TFIIH on DNA repair.
The XPC protein participates in general DNA repair: cells mutated on the XPC gene carry out normal transcription coupled repair, but are defective in overall repair (van Hoffen et al., 1995). Results of in vitro experiments indicate that the presence of XPC protein is not fundamental for repair (Mu et al., 1995), but in this circumstance both the excised oligomer and the damaged strand in the pre-incision complex are extensively degraded. This protein also associates with the TFIIH (Drapkin et al., 1994) and has a high nonspecific DNA affinity. These data lead to the reasonable assumption that XPC can help stabilize the pre-incision subassemblies on nucleosomal DNA, while protecting the rest of the DNA. In transcribed DNA, an elongation complex stalled at a lesion obviates the need for XPC.
Once the damage is recognized and prepared for repair, the DNA is nicked in both sides of the lesion. The XPF and the XPG proteins are implicated in this process: the incision at 5of the lesion is made by the XPF protein (complexed with the ERCC1 protein) and at 3 by the XPG protein (Matsunaga et al., 1995). After the incision and excision of the damaged DNA strand, the synthesis of the" new" DNA by DNA polymerases d or e occurs and the final nick is removed by DNA ligase.
The second human nucleotide excision repair disease is Cockayne syndrome (CS). CS patients are also sun sensitive but show a distinctive array of congenital neurological and skeletal abnormalities, including mental deficiency and dwarfism (Nance and Berry, 1992). Cells from these patients are very sensitive to UV irradiation and have a defective RNA synthesis after UV. Mutations in CSA and CSB genes are responsible for the classical CS. However, the symptoms of the disease are also seen in rare XP patients belonging to the groups B, D and G (Vermeulen et al., 1993).
The real function of CSA and CSB genes are not known, yet. However, there are some findings that associate these genes with the preferential repair of transcribed genes. Cells from patients with CS are defective in preferential repair (Friedberg, 1996) and the sequence of CSB gene reveal some similarity to the Escherichia coli Mfd gene, a transcription repair coupling factor (TRFC) (Troelstra et al., 1992). The homology in the ATPase/helicase motif is high and this motif is essential for the function of the Mfd gene. These observations have led to the suggestion that CSB protein, complexed with the CSA protein, may function as TRFC in human cells. The following model is proposed for the action of these proteins: the CSB-CSA heterodimer recognizes the RNA polymerase II stalled at a lesion, backs off this polymerase without dissociating the ternary complex, and then the heterodimer recruits XPA and TFIIH to the lesion site, necessary for the repair of transcribed genes.
The third disease in this group is trichothiodystrophy (TTD) whose main symptoms are brittle hair and nails (because of a reduced content of cysteine-rich matrix proteins), ichthyotic skin, and physical and mental retardation (Stefanini et al., 1993). Some of the TTD patients show photosensitivity, correlated with a defective excision repair, but no cases of skin cancer have been related. The TTD are caused by mutations in three genes: TTD-A, XPB and XPD. Likewise CS, the TTD individuals with mutations in XPD may also have XP syndromes. The TTD is mainly a transcription disease, because the deficiency of these three groups can be restored by the micro-injection of the TFIIH factor, though none of the known TFIIH subunits is mutated in TTD-A gene (Sancar, 1996).
The fact that these three different diseases (XP, CS and TTD) may be mutated in the same genes (as is the case of XPB, XPD and XPG) is intriguing. The clinical symptoms of these genetic disorders are distinct specially concerning the increased frequency of skin tumors, observed in XP but not in CS or TTD patients. These different symptoms may simply express differences in the abilities of the mutated cells to perform either DNA repair or transcription, depending on the mutation on the gene. However, recent data have shone some light onto this very interesting question. Working with cells derived from XPG patients associated or not with CS, Cooper et al. (1997) have found evidence that the developmental CS defects may be due to a defective preferential repair of active genes by oxidative damage. On the other hand, Ahrens et al. (1997) have found that the high risk of cancer in XPD patients may be due to a decreased immunological response to UVB irradiation, not found on cells from TTD patients (mutated in the same XPD gene). The author propose that the XPD protein might have a transcriptional role controlling the expression of immunological relevant genes. Thus, mutations that affect this control (such as those found in XP patients, but not in TTD) may increase the skin cancer risk.
Other human syndromes possibly linked to DNA repair defects
Additional human genetic disorders have features of genome instability that may be linked to defective DNA repair or at least to the processing of DNA: Fanconis anemia (FA), Bloom syndrome (BS) and ataxia telangiectasia (AT). Patients with these syndromes have increased number of spontaneous chromosomal aberrations in their cells and there is evidence for increased frequency of development of tumors early in life (Table II).
*These genes are members of the gene family of RecQ helicases.
Fanconis anemia patients have decreased number of all cellular elements of the blood (i.e., erythrocytes, leukocytes and platelets), hyperpigmentation and short stature. Cultured cells from some FA patients are sensitive to chemical agents, such as mitomycin C (MMC) or photoactivated psoralen derivatives, that induce the formation of interstrand crosslinks on DNA. Thus, FA cells seem to have a defect on the specific repair for this kind of lesions. However, this is not true for all FA patients, indicating certain genetic heterogeneity for the disease. In fact, at least five genetic complementation groups (groups A to E) have been found associated with FA. Only two of the genes have been cloned, FAA (Lo Ten Foe et al., 1996) and FAC (Strathdee et al., 1992), but no specific functions have been attributed to the FAA and FAC proteins until now. The cDNA clone for the FAA gene encodes a polypeptide predicted to contain 2 overlapping bipartite nuclear localization signals and a partial leucine zipper consensus sequence, suggesting that the protein is localized in the nucleus. However, the FAC protein is normally found in the cytoplasm (Yamashita et al., 1994), which is not expected for a DNA repair enzyme. Recent data reveal that cells from FA patients also have elevated recombination activity (Thyagarajan and Campbell, 1997) and deregulated apoptosis (Ridet et al., 1997). These findings implicate that the FA genes may play major roles in the control of DNA metabolism and of apoptosis.
The syndrome initially described by Bloom (1954) is characterized by pre- and postnatal growth deficiency, sunlight sensitivity, hypo- and hyper-pigmented skin, predisposition to malignancy and chromosomal instability. Light-induced telangiectasia develops typical "butterfly" lesions on the patients face. Cells from patients with BS grow slowly in culture, with low plating efficiencies and long generation times. DNA synthesis is unpaired, with an accumulation of replication intermediates (Giannelli et al., 1977), suggesting a defect associated with DNA replication, although this is not yet understood. The genetic mapping of BS related families allowed the cloning of the gene responsible for this disease, denominated BLM (Ellis et al., 1995). Strikingly, this gene has homology with a family of RecQ helicases, named after the Escherichia coli RecQ gene. In bacteria this gene is a member of the RecF recombination pathway, involved in conjugational recombination proficiency and UV resistance. Ellis et al. (1995) suggested that the absence of the BLM gene product probably destabilizes other enzymes that participate in DNA replication and repair, perhaps through direct interactions and through more general responses to DNA damage. More recently, a second human gene member of the RecQ family was identified, WRN, and defects on this gene may lead to the Werner syndrome (WS) (Yu et al., 1996). The main clinical feature of this syndrome is premature aging, but many other features (such as short stature, neoplasia, hyperrecombination, cells with defects on DNA initiation and chain elongation) resemble those of BS. Although DNA repair seems to be normal in the cells from BS and WS, the BLM and WRN proteins are implicated in the DNA metabolism. They probably participate in a large family of RecQ helicases in the human genome, important to maintain genomic stability.
Finally, ataxia telangiectasia (AT) is another autosomal recessive disorder characterized by cerebellar ataxia, progressive mental retardation, immune defects, severe muscular incoordination, and a strong predisposition to malignancy, particularly in the lymphoreticular system. The term telangiectasia describes the dilatation of blood vessels, particularly in the eyes and skin. Chromosomal breakage is also a common feature. AT cells are abnormally sensitive to killing by ionizing radiation and for long it has been considered an X-ray analogue of XP, since XP patients are sensitive to UV light and not X-ray. However, unlike the observations with XP, no clearly defect on DNA repair was identified in AT cells. Curiously, the AT cells are abnormally resistant to the inhibition of DNA synthesis by ionizing radiation, suggesting a defect in the processing of DNA damage. The latter trait has been used to identify 4 complementation groups for the classical form of the disease (Jaspers et al., 1988), which, however, are all associated with mutations on the same recently cloned gene, named ATM (for AT mutated, Savitsky et al., 1995). The ATM gene encoded a putative protein that is homolog to several yeast and mammalian phosphatidylinositol 3-prime (PI-3) kinases, that are involved in mitogenic signal transduction, meiotic recombination, and cell cycle control. This is consistent with cell cycle defects observed in AT cells, including the absence of G1-S checkpoint after ionizing radiation (Kastan et al., 1992).
Although, as for the other genetic disorders described above, AT is a very rare disease, affecting about 1:40,000 live births, the cloning of the ATM gene has a great social impact. The disease is found only in individuals with mutations in both alleles, that is, in homozygosis. Heterozygous relatives of AT patients are apparently normal, but there are indications that they are more likely to develop tumors than individuals unrelated to these patients. Since the heterozygous individuals are very frequent in the population, about 1%, it has been estimated that heterozygosis of the AT disease may be associated with an important fraction of persons that die of cancer before the age of 45 (Swift et al., 1987). This is particularly clear in association with breast cancer: approximately 8.8% of the patients with breast cancer in the US white population might be heterozygous for the ATM gene (Swift et al., 1987). The identification of the most frequent mutations that inactivate this gene will provide an important tool for the screening and diagnosis of cancer-prone individuals.
Human DNA repair and cancer associated diseases
A different cause for genetic instability is the frequent appearance of mismatches on the double helix of DNA. Base-base mismatches and small loops may occur during most of the main mechanism that process DNA, that is, replication, recombination and repair itself. From bacteria to eukaryotes, there are special enzymatic machineries that correct these mismatches, restoring the original DNA sequence, which are generally called DNA mismatch repair. In Figure 2, a scheme of the DNA mismatch repair is presented.
Defects on DNA repair in bacteria have long been known as a cause for a mutator phenotype. Thus, such a phenotype would be also expected in human cells, and it could also be related to hereditary cancer. In fact, one of the most interesting recent findings in the field was the discovery that mutations in mismatch repair genes (such as hMSH2, hMLH1, hPMS1 and hPMS2) are responsible for the hereditary nonpolyposis colorectal cancer (HNPCC), which affects as many as 1 in 200 individuals, and also a subset of sporadic colorectal cancers (Jiricny, 1994; Eshleman and Markowitz, 1995). Individuals are normally heterozygous for the mismatch genes, but the loss of heterozygosity, that may occur spontaneously, leads to genetic instability, originating tumor cells. In fact, cells from tumors of HNPCC patients show increased microsatellite instability and are normally defective for DNA mismatch repair. As for the ATM gene, the availability of the DNA mismatch repair gene sequences will aid the screening of HNPCC families for the most frequent mutated alleles. Moreover, other types of sporadic cancers are found to be associated with the instability of microsatellite DNA sequences, probably due to a deficient mismatch repair.
It seems clear that new genes involved with DNA repair will be found in humans with potential connection with cancer. This seems to be the case of two genes that are associated with breast cancer susceptibility (BRCA1 and BRCA2). These two genes were cloned recently (Szabo et al., 1996; Tavtigian et al., 1996) linked to dominant inheritance of familial breast cancer: mutations in one of these genes somehow increase enormously the frequency of tumor on the individuals. The mechanisms of action are still not completely understood. The fact that two proteins do not resemble anything in existing databases does not give any clue on their functions. However, recent work by Sharan and colleagues (1997) indicated that BRCA2 protein plays a critical role in DNA repair. They observed that BRCA2 protein is able to bind to the human homologue of the yeast RAD51 protein. In yeast this protein participates in the DNA repair of double strand break and recombination. Consistent with this observation, embryonic mouse cells, in which the murine version of BRCA2 has been inactivated, are very sensitive to ionizing radiation. The BRCA1 protein also has the capacity to bind to the human RAD51 protein (Scully et al., 1997) and it may also participate in the same DNA repair pathway.
Conclusions and perspectives
The vigorous efforts to isolate and characterize DNA repair genes have helped to understand some aspects of the cell mechanisms for DNA damage processing and their involvement with human syndromes and cancer formation. It is now clear that there are strong connections among the different enzymatic machineries that handle nucleic acids within the cell. The network that connects DNA repair with DNA replication, transcription, cell cycle control, apoptosis, etc., is the guardian of genome stability. Failure of one of these processes can cause mutations and, consequently, cancer. In fact, the notion that genome instability may generate tumors has been known for a long time. Thus, the discoveries reported above are not surprising: those gene products that look after the cell genome, eliminating DNA lesions and errors of the DNA metabolism (that is, DNA repair proteins), must be operational, otherwise, there will be increased tumor risk. However, the list of genes presented in this work seems to be only the tip of an iceberg. The complexity of DNA repair mechanisms is far from being elucidated, and certainly a larger number of genes is involved in nuclear genome stability. The identification of such genes will contribute not only to our knowledge of the cellular and gene metabolism, but also to cancer prevention.
ACKNOWLEDGMENTS
C.R.M. is the recipient of a post-doctoral fellowship from FAPEMIG (Minas Gerais, Brazil) and this work was supported by CNPq (Brasília, Brazil) and FAPESP (São Paulo, Brazil). Publication supported by FAPESP.
- Ahrens, C., Grewe, M., Berneburg, M., Grether-Beck, S., Quilliet, X., Mezzina, M., Sarasin, A., Lehmann, A.R., Arlett, C.F and Krutmann, J. (1997). Photocarcinogenesis and inhibition of intercellular adhesion molecule 1 expression in cells of DNA-repair-defective individuals. Proc. Natl. Acad. Sci. USA 94: 6837-6841.
- Bloom, D. (1954). Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs. Am. J. Dis. Child 88: 754-758.
- Bohr, V.A., Smith, C.A., Okumoto, D.S and Hanawalt, P.C. (1985). DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40: 359-369.
- Bootsma, D. and Hoeijmakers, J.H.J. (1991). The genetic basis of xeroderma pigmentosum. Ann. Genet. 34: 143-150.
- Cleaver, J.E. (1968). Defective repair replication of DNA in xeroderma pigmentosum. Nature 218: 652-656.
- Cooper, P.K., Nouspikel, T., Clarkson, S.G. and Leadon, S.A. (1997). Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science 275: 990-993.
- Drapkin, R., Reardon, J.T., Ansari, A., Huang, J.C., Zawel, L., Ahn, K., Sancar, A. and Reinberg, D. (1994). Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature 368: 769-772.
- Ellis, N.A., Groden, J., Ye, T.Z., Straughen, J., Lennon, D.J., Ciocci, S., Proytcheva, M. and German, J. (1995). The Bloom syndrome gene product is homologous to RecQ helicases. Cell 83: 655-666.
- Eshleman, J.R. and Markowitz, S.D. (1995). Microsatellite instability in inherited and sporadic neoplasms. Curr. Opin. Oncol. 7: 83-89.
- Friedberg, E.C. (1996). Relationships between DNA repair and transcription. Annu. Rev. Biochem. 65: 15-42.
- Giannelli, F., Benson, P.F., Pawsey, S.A. and Polani, P.E. (1977). Ultraviolet light sensitivity and delayed DNA-chain maturation in Bloom syndrome fibroblasts. Nature 265: 466-469.
- Goodrich, J.A. and Tjian, R. (1994). Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell 77: 145-156.
- He, Z., Henricksen, L.A., Wold, M.S. and Ingles, C.J. (1995). RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 374: 566-569.
- Jaspers, N.G.J., Gatti, R.A., Baan, C., Linssen, P.C.M.L. and Bootsma, D. (1988). Genetic complementation analysis of Ataxia Telangiectasia and Nijmegen breakage syndrome: a survey of 50 patients. Cytogenet. Cell Genet. 49: 259-263.
- Jiricny, J. (1994). Colon cancer and DNA repair: have mismatches met their match? Trends Genet. 10: 164-168.
- Kastan, M.B., Zhan, Q., El-Deiry, W.S., Carrier, F., Jacks, T., Walsh, W.V., Plunkett, B.S., Vogelstein, B. and Fornace Jr., A.J. (1992). A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71: 587-597.
- Li, L., Lu, X., Peterson, C.A. and Legerski, R.J. (1995). An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol. Cell. Biol. 15: 5396-5402.
- Lo Ten Foe, J.R., Rooimans, M.A., Bosnoyan-Collins, L., Alon, N., Wijker, M., Parker, L., Lightfoot, J., Carreau, M., Callen, D.F., Savoia, A., Cheng, N.C., van Berkel, C.G.M., Strunk, M.H.P., Gille, J.J.P., Pals, G., Kruyt, F.A.E., Pronk, J.C., Arwert, F., Buchwald, M. and Joenje, H. (1996). Expression cloning of a cDNA for the major Fanconis anaemia gene, FAA. Nat. Genet. 14: 320-323.
- Matsunaga, T., Mu, D., Park, C.H., Reardon, J.T. and Sancar, A. (1995). Human DNA repair excision nuclease. Analysis of the roles of the subunits involved in dual incisions by using anti-XPG and anti-ERCC1 antibodies. J. Biol. Chem. 270: 20862-20869.
- Mu, D., Park, C.H., Matsunaga, T., Hsu, D.S., Reardon, J.T. and Sancar, A. (1995). Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270: 2415-2418.
- Nance, M.A. and Berry, A.S. (1992). Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 42: 68-84.
- Park, C.H. and Sancar, A. (1994). Formation of a ternary complex by human XPA, ERCC1, and ERCC4(XPF) excision repair proteins. Proc. Natl. Acad. Sci. USA 91: 5017-5021.
- Park, C.H., Mu, D., Reardon, J.T. and Sancar, A. (1995). The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J. Biol. Chem. 270: 4896-4902.
- Ridet, A., Guillouf, C., Duchaud, E., Cundari, E., Fiore, M., Moustacchi, E. and Rosseli, F. (1997). Deregulated apoptosis is a hallmark of the Fanconis anemia syndrome. Cancer Res. 57: 1722-1730.
- Sancar, A. (1996). DNA excision repair. Annu. Rev. Biochem. 65: 43-81.
- Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D.A., Smith, S., Uziel, T., Sfez, S., Ashkenazi, M., Pecker, I., Frydman, M., Harnik, R., Patanjali, S.R., Simmons, A., Clines, G.A., Sartiel, A., Gatti, R.A., Chessa, L., Sanal, O., Lavin, M.F., Jaspers, N.G.J., Taylor, A.M.R., Arlett, C.F., Miki, T., Weissman, S.M., Lovett, M., Collins, F.S. and Shilon, Y. (1995). A single Ataxia Telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749-1753.
- Scully, R., Chen, J., Plug, A., Xiao, Y., Weaver, D., Feunteun, J., Ashley, T. and Livingston, D.M. (1997). Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265-275.
- Sharan, S.K., Morimatsu, M., Albrecht, U., Lim, D.S., Regel, E., Dinh, C., Sands, A., Eichele, G., Hasty, P. and Bradley, A. (1997). Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386: 804-810.
- Stefanini, M., Vermeulen, W., Weeda, G., Giliani, S., Nardo, T., Mezzina, M., Sarasin, A., Harper, J.I., Arlett, C.F., Hoeijmakers, J.H. and Lehmann, A.R. (1993). A new nucleotide-excision-repair gene associated with the disorder trichothiodystrophy. Am. J. Hum. Genet. 53: 817-821.
- Strathdee, C.A., Gavish, H., Shannon, W.R. and Buchwald, M. (1992). Cloning of a cDNAs for Fanconis anemia by functional complementation. Nature 356: 763-767.
- Swift, M., Reitnauer, P.J., Morrell, D. and Chase, C.L. (1987). Breast and other cancers in families with ataxia-telangiectasia. N. Engl. J. Med. 316: 1289-1294.
- Szabo, C.I., Wagner, L.A., Francisco, L.V., Roach, J.C., Argonza, R., King, M.C. and Ostrander, E.A. (1996). Human, canine and murine BRCA1 genes: sequence comparison among species. Hum. Mol. Genet. 5: 1289-1298.
- Tavtigian, S.V., Simard, J., Rommens, J., Couch, F., Shattuck-Eidens, D., Neuhausen, S., Merajver, S., Thorlacius, S., Offit, K., Stoppa-Lyonnet, D., Belanger, C., Bell, R., Berry, S., Bogden, R., Chen, Q., Davis, T., Dumont, M., Frye, C., Hattier, T., Jammulapati, S., Janecki, T., Jiang, P., Kehrer, R., Leblanc, J.F., Mitchel, J.T., McArthur-Morrison, J., Nguyen, K., Peng, Y., Samson, C., Schroeder, M., Snyder, S.C., Steele, L., Stringfellow, M., Stroup, C., Swedlund, B., Swensen, J., Teng, D., Thomas, A., Tran, T., Tranchant, M., Weaver-Feldhaus, J., Wong, A.K.C., Shizuya, H., Eyfjord, J.E., Cannon-Albright, L., Labrie, F., Skolnick, M.H., Weber, B., Kamb, A. and Goldgar, D.E. (1996). The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat. Genet. 12: 333-337.
- Thyagarajan, B. and Campbell, C. (1997). Elevated homologous recombination activity in Fanconis anemia fibroblasts. J. Biol. Chem. 272: 23328-23333.
- Troelstra, C., van Gool, A., de Wit, J., Vermeulen, W., Bootsma, D. and Hoeijmakers, J.H.J. (1992). ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne syndrome and preferential repair of active genes. Cell 71: 939-953.
- van Hoffen, A., Venema, J., Meschini, R., van Zeeland, A.A. and Mullenders, H.F. (1995). Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in Xeroderma Pigmentosum group C fibroblasts. EMBO J. 14: 360-367.
- Vermeulen, W., Jaeken, J., Jaspers, N.G., Bootsma, D. and Hoeijmakers, J.H.J. (1993). Xeroderma pigmentosum complementation group G associated with Cockayne syndrome. Am. J. Hum. Genet. 53: 185-192.
- Yamashita, T., Barber, D.L., Zhu, Y., Wu, N. and DAndrea, A.D. (1994). The Fanconis anemia polypeptide FACC is localized to the cytoplasm. Proc. Natl. Acad. Sci. USA 91: 6712-6716.
- Yu, C.E., Oshima, J., Fu, Y.H., Wijsman, E.M., Hisama, F., Alisch, R., Matthews, S., Nakura, J., Miki, T., Ouais, S., Martin, G.M., Mulligan, J. and Schellenberg, G.D. (1996). Positional cloning of the Werner syndrome gene. Science 272: 258-262.
Publication Dates
-
Publication in this collection
06 Oct 1998 -
Date of issue
Dec 1997