Abstract
The equilibrium unfolding of bovine trypsinogen was studied by circular dichroism, differential spectra and size exclusion HPLC. The change in free energy of denaturation was <img SRC="http:/img/fbpe/bjmbr/v32n6/3282sup.gif" ALIGN="BOTTOM" BORDER="0" VSPACE="0" HSPACE="0"> = 6.99 ± 1.40 kcal/mol for guanidine hydrochloride and <img SRC="http:/img/fbpe/bjmbr/v32n6/3282sup.gif" ALIGN="BOTTOM" BORDER="0" VSPACE="0" HSPACE="0"> = 6.37 ± 0.57 kcal/mol for urea. Satisfactory fits of equilibrium unfolding transitions required a three-state model involving an intermediate in addition to the native and unfolded forms. Size exclusion HPLC allowed the detection of an intermediate population of trypsinogen whose Stokes radii varied from 24.1 ± 0.4 Å to 26.0 ± 0.3 Å for 1.5 M and 2.5 M guanidine hydrochloride, respectively. During urea denaturation, the range of Stokes radii varied from 23.9 ± 0.3 Å to 25.7 ± 0.6 Å for 4.0 M and 6.0 M urea, respectively. Maximal intrinsic fluorescence was observed at about 3.8 M urea with 8-aniline-1-naphthalene sulfonate (ANS) binding. These experimental data indicate that the unfolding of bovine trypsinogen is not a simple transition and suggest that the equilibrium intermediate population comprises one intermediate that may be characterized as a molten globule. To obtain further insight by studying intermediates representing different stages of unfolding, we hope to gain a better understanding of the complex interrelations between protein conformation and energetics.
trypsinogen; protein denaturation; thermodynamic stability; intermediates; molten globule
Braz J Med Biol Res, June 1999, Volume 32(6) 673-682
Partially folded intermediates during trypsinogen denaturation
N.F. Martins and M.M. Santoro
Laboratório de Enzimologia e Físico-Química de Proteínas, Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brasil
References

The equilibrium unfolding of bovine trypsinogen was studied by circular dichroism, differential spectra and size exclusion HPLC. The change in free energy of denaturation was  = 6.99 ± 1.40 kcal/mol for guanidine hydrochloride and = 6.37 ± 0.57 kcal/mol for urea. Satisfactory fits of equilibrium unfolding transitions required a three-state model involving an intermediate in addition to the native and unfolded forms. Size exclusion HPLC allowed the detection of an intermediate population of trypsinogen whose Stokes radii varied from 24.1 ± 0.4 Å to 26.0 ± 0.3 Å for 1.5 M and 2.5 M guanidine hydrochloride, respectively. During urea denaturation, the range of Stokes radii varied from 23.9 ± 0.3 Å to 25.7 ± 0.6 Å for 4.0 M and 6.0 M urea, respectively. Maximal intrinsic fluorescence was observed at about 3.8 M urea with 8-aniline-1-naphthalene sulfonate (ANS) binding. These experimental data indicate that the unfolding of bovine trypsinogen is not a simple transition and suggest that the equilibrium intermediate population comprises one intermediate that may be characterized as a molten globule. To obtain further insight by studying intermediates representing different stages of unfolding, we hope to gain a better understanding of the complex interrelations between protein conformation and energetics.
= 6.99 ± 1.40 kcal/mol for guanidine hydrochloride and = 6.37 ± 0.57 kcal/mol for urea. Satisfactory fits of equilibrium unfolding transitions required a three-state model involving an intermediate in addition to the native and unfolded forms. Size exclusion HPLC allowed the detection of an intermediate population of trypsinogen whose Stokes radii varied from 24.1 ± 0.4 Å to 26.0 ± 0.3 Å for 1.5 M and 2.5 M guanidine hydrochloride, respectively. During urea denaturation, the range of Stokes radii varied from 23.9 ± 0.3 Å to 25.7 ± 0.6 Å for 4.0 M and 6.0 M urea, respectively. Maximal intrinsic fluorescence was observed at about 3.8 M urea with 8-aniline-1-naphthalene sulfonate (ANS) binding. These experimental data indicate that the unfolding of bovine trypsinogen is not a simple transition and suggest that the equilibrium intermediate population comprises one intermediate that may be characterized as a molten globule. To obtain further insight by studying intermediates representing different stages of unfolding, we hope to gain a better understanding of the complex interrelations between protein conformation and energetics.
Key words: trypsinogen, protein denaturation, thermodynamic stability, intermediates, molten globule
Abstract
Introduction
Protein folding was generally believed to be a highly cooperative two-state process in which only the native and completely unfolded states were significantly populated with no apparent intermediates. Of special interest in understanding the factors that determine the three-dimensional structure of globular proteins is the detection and identification of intermediates in folding reactions, since recent equilibrium and kinetic studies have shown the existence of stable intermediate conformational states for several proteins (1,2). One approach to this problem is the study of equilibrium denaturation of the proteins since it is not yet possible to predict the folded conformation of a protein from its sequence alone.
The intermediates that define a folding mechanism are likely to contain some regions with relatively well-defined conformation and other regions that are highly disordered. The partially folded intermediates have a wide distribution from a high ordered molten globule (MG) (close to the native state) to a pre-molten globule (close to the unfolded state), with solvent-accessible hydrophobic regions (3,4). By definition, a molten globule state would share a compact globular structure with the native conformation. Also, it would have an accessible hydrophobic core with increased fluctuations of side chains, a nearly native secondary structure and almost no tertiary structure. The MG state was first described for the proteins a-lactalbumin (5) and cytochrome c (6). Investigation of a-lactalbumin revealed that under certain denaturing conditions a stable intermediate state was formed which could be distinguished from the native and unfolded state. This intermediate was demonstrable as non-coincident denaturation transition when monitored by near and far ultraviolet circular dichroism (UV CD). Later kinetic experiments demonstrated for several other proteins that denaturation can occur through another compact intermediate, named pre-molten globule (7,8). This state has a substantial amount of fluctuating secondary structure, is partly compact and has solvent-accessible non-polar clusters. This evidence suggests that the molten globule and other intermediates are obligatory in the folding pathway, although their exact significance in the mechanism of folding is still not understood (9).
Trypsinogen is an interesting model for folding studies; it is structurally very similar to trypsin with the advantage of a very low proteolytic activity. Trypsinogen is a globular protein with 229-amino acid residues and six disulfide bridges. The structure has two similar domains with a predominance of ß strands and its activation domain (33 hydrophilic amino acids in the N-terminal sequence) is rather disordered compared to the active enzyme (10).
Bulaj and Otlewsky (11,12) previously studied the denaturation of trypsinogen at pH 5.8 by both thermal and chemical procedures. The authors showed that the stability of trypsinogen is increased by calcium ion, by the dipeptide Ile-Val and by basic pancreatic trypsin inhibitor (BPTI) and proposed a two-state transition in equilibrium. The unfolding kinetics of trypsinogen was studied by Otlewski et al. (13) using a fluorescence-detected stopped-flow manifesting two independent phases. The fast phase is not a single-phase event under mildly denaturing conditions and in the slow phase the authors showed evidence of intermediates accumulating under strongly folding conditions. A similar kinetics was observed for the trypsinogen-Ca2+ complex and limited data on trypsin unfolding show a virtually identical behavior (13). More recently Ruan et al. (14) studied the effect of hydrostatic pressure on the unfolding of bovine trypsin. These authors showed a stable partly denatured state at 6.5 kbar having characteristics of a molten globule state.
In this present study we report the equilibrium denaturation of bovine trypsinogen in both guanidine hydrochloride (GdnHCl) and urea at pH 7.0 and compare the spectroscopic and hydrodynamic properties of the protein during the unfolding process. We demonstrate the presence of an intermediate equilibrium state which could be a molten globule and provide experimental evidence for other intermediates in the unfolding pathway.
Material and Methods
Reagents and buffers
The buffers used for the differential spectroscopy, near UV CD and size exclusion studies contained 100 mM 2-(4-morpholine) ethanesulfonic acid (MES), pH 7.0, 20 mM CaCl2 and 81 mM NaNO3. Unfolding conditions were achieved by appropriate additions of 10 M ultrapure urea or 8 M GdnHCl previously purified in the same buffers. The concentration of GdnHCl and urea solutions was confirmed by refractive index measurement (15). All proteins were from Sigma Chemical Co., St. Louis, MO, USA, and the other reagents were of analytical grade.
Purification of commercial bovine trypsinogen
The purification procedure was performed on an ion exchange column (DEAE-Trisacryl - 1.0 x 30 cm), where 50 mg of the sample was loaded in the system and eluted with 10 mM Tris, pH 7.1, 2 mM CaCl2, 10 mM NaCl and 1 mM benzamidine at a flow rate of 9.6 ml/h. The effluent was monitored by absorption at 280 nm and tested for amidase activity according to the method of Ascenzi et al. (16). Trypsinogen was recovered in the major protein peak with no amidase activity. The effluent was dialyzed in 1 mM HCl for 4 h at 4oC and freeze-dried. The peak fraction was submitted to HPLC on a reverse phase analytical column (Novapak C-18 - 3.9 x 20 cm - Waters-Millipore). The column was eluted with 1% TFA (v/v) in an acetonitrile gradient from 0 to 60% (v/v) at a flow rate of 60 ml/h. The same sample was submitted to SDS-polyacrylamide gel electrophoresis by the method of Laemmli (17). The purified fraction presented a single band by SDS gel electrophoresis and a single peak by reverse phase-HPLC in a Novapack column. The N-terminal sequence of this fraction was Val-(Asp)4-Lys-Ile-Val (18), determined by the method described by Chang (19).
Sample concentration
Sample concentrations in stock solutions were determined by UV measurements recorded with a HITACHI UV 160 A spectrophotometer using x = 33360 M-1 cm-1 as the molar absorbance coefficient at 280 nm (11). The protein concentration in each sample was 1 mM for near-UV CD, differential spectroscopy (DS) and size exclusion (SE)-HPLC experiments, and 250 µM for far-UV analysis. To prevent autoactivation, 1 mol of a-1-antitrypsin was added to 100 mol of trypsinogen solutions (11). All solutions were filtered through a 0.45-µm Millipore filter before the measurements.
Denaturation experiments
For the denaturation experiments, protein samples were incubated for 18 h at each GdnHCl or urea concentration in buffer prior to the spectroscopic measurements.
Differential spectroscopy
The GdnHCl and urea unfolding curves were obtained by measuring the difference in UV signal at 293 nm (DS), at 25oC, with a HITACHI UV 160 A spectrophotometer with twin cells with a 0.45-cm path length. The protein concentration was 1 mM for all measurements. The instrument was calibrated with a potassium dichromate solution.
Circular dichroism
CD studies were done using a Jasco J20 spectropolarimeter. The tertiary structure was monitored in the near-UV region (250-350-nm wavelength range) using a cell with a 0.5-cm path length. Protein concentration was 1 mM. All measurements were made at 25oC. The instrument was calibrated with d-camphor sulfonic acid using a molar ellipticity of 7800 degrees cm2 dmol-1 at 290.5 nm. The buffer blanks were subtracted from each CD spectrum. Since it has been shown that CD spectra between 260 and approximately 180 nm can be analyzed for the different secondary structural types, i.e., alpha helix, parallel and antiparallel beta sheet, turn and others (20), the secondary structure was monitored in the far-UV region. The data were collected with a Jobin-Yvon spectropolarimeter at 222 nm wavelength, using a cell of 0.1-cm path length, at 25oC. Samples were prepared at a concentration of 250 µM in 20 mM phosphate buffer, pH 7.0, and incubated for 18 h before the measurements.
Stokes radius - SE-HPLC
Stokes radii (Rs) for the native, intermediate and denatured states were determined by size-exclusion HPLC. Protein concentration was 1 mM as determined spectrophotometrically. The determinations were carried out with a Shodex - Protein KW 803 apparatus with dimensions of 0.8 x 30 cm (Showa Denko & Shoko, Tokyo, Japan), connected to a Shimadzu HPLC system equipped with LC10-AD pumps, an SPD 10-A UV detector (set at 280 nm) and a CR4-A integrator. The column was calibrated with a-lactalbumin, carbonic anhydrase, egg albumin, bovine albumin and urease. Stokes radii were determined as described by Uversky (21).
1,8 ANS binding
An Aminco-Bowman spectrofluorometer was used for experiments involving the binding of 1,8 aniline-naphthalene sulfonate (ANS) to trypsinogen. Excitation was at 420 nm and relative emission intensity was monitored at 480 nm. The excitation and emission bandwidths were fixed at 2 nm each and a 1-cm path length quartz cuvette was used for all measurements. The samples were incubated for 15 min (22). The spectrum of each sample was corrected by subtracting the buffer. Protein concentration was 500 µM for all assays.
Analysis of unfolding curves
The unfolding curves induced by guanidine hydrochloride or urea monitored by different techniques were normalized for the apparent fraction of unfolded protein (fU) versus denaturant concentration. fU was calculated from the data by the following equation:

where YO is the observed value of the spectroscopic parameter at a given denaturant concentration and YN and YU are the values of the native and unfolded baselines at the same denaturant concentration. The linear fit was calculated by the methods of Pace (23) and Santoro and Bolen (24), which describe a two-state transition between the native and the unfolded states. The fit for three-state transition was calculated according to the equation described in Matthews and Crisanti (25):

where I is a partially unfolded form of the protein. Then:

where Kapp is the overall apparent equilibrium constant, and K1 and K2 are constants for each step. The Z parameter is the fractional change in the optical parameters in the transition from N to I and is defined by:

where YI is the value of a given optical parameter for the intermediate species, I.
Statistical analysis
The statistical F test was applied according to the equation proposed by Izquierdo et al. (26) in order to discriminate between the fits of different degree, i.e., fits with different numbers of parameters:

where Q1 and Q2 are the sum of squared residuals for models of two-state transition and three-state transition, respectively, p1 and p2 refer to the corresponding number of parameters, and n refers to the number of experimental data. A 95% confidence level was used for each F value determined in this way, allowing us to identify the minimum degree that fitted the data.
Equilibrium unfolding of trypsinogen induced by GdnHCl
Figure 1 and Table 1 show the denaturation of trypsinogen at pH 7.0 and 25oC using GdnHCl as denaturant, determined by two techniques at three different wavelengths, differential spectra (DS) at 293 nm and circular dichroism ([Q]) at 300 nm and at 222 nm. The unfolding of trypsinogen monitored by changes in the microenvironment of aromatic residues (DS at 293 nm and CD at 300 nm) is described as a two-state transition. Since these two results show coincident transitions, the data of DS at 293 nm and CD at 300 nm were combined to calculate the free energy according to a two-state model and protein stability was estimated as 7.72 kcal/mol.
- Unfolding transition of trypsinogen in 100 mM MES, pH 7.0, 20 mM CaCl2 and 81 mM NaNO3, pH 7.0, as a function of GdnHCl concentration. The fraction of unfolded protein (fU) was calculated from the ratio of differential spectroscopy (DS) signal at 293 nm (triangles) and molar ellipticity ([Q]) at 300 nm (black circles) and at 222 nm (open circles). The fit for a two-state transition is represented by the dashed line for DS 293 nm and CD 300 nm and by the dotted line for CD 222 nm. The fits were calculated according to Santoro and Bolen (24). The three-state fit of CD at 222 nm (solid line) was calculated according to Matthews and Crisanti (25). The near-UV CD was monitored at 300 nm using a 0.5-cm path length cuvette. The spectroscopic measurements were made in a 0.5-cm path length tandem cell as described in the procedures. Protein concentration was 1 mM for each sample. The far-UV CD was monitored at 222 nm using a 0.1-cm path length cuvette. Protein concentration was 250 µM and temperature 25oC.
On the other hand, the unfolding transition of trypsinogen monitored by CD at 222 nm fitted better a three-state model, with a total = 6.69 kcal/mol and an intermediate state accumulating at 2.8 M denaturant concentration. The selection of the model to fit the data was based on an F test, as described previously. The value of F(p2 - p1, n - p2) was 8.63, taken from the statistical table for the F test at the 0.95 confidence level. The calculated F value, based on the c2 of the experimental data, was F = 41.1, indicating that the three-state model is a better model to fit these data (25). These results suggest that the equilibrium curve should be represented as a composite of at least two transitions between the native and intermediate state and between the intermediate and the denatured state. Also, the non-coincidence of far-UV CD and near-UV CD suggests the presence of intermediate equilibrium states.
Size exclusion chromatography in GdnHCl and Stokes radius calculation
Since the curve monitored by CD at 222 nm indicated a complex transition, we used SE-HPLC as an appropriate technique to identify intermediate states. The elution profiles of GdnHCl-induced denaturation of trypsinogen at 25oC are presented in Figure 2. The Stokes radii of native and unfolded states were calculated to be 19.8 ± 0.3 Å and 28.1 ± 0.2 Å, respectively. In solvent-induced unfolding the hydrodynamic volume of the protein molecule also changed, showing an intermediate state in the range of the transition region at a concentration of 1.5 M GdnHCl. The Stokes radius for this intermediate fraction was 24.1 ± 0.4 Å. As the concentration of the denaturant increased, the fraction changed in retention time to a peak corresponding to a Stokes radius calculated to be 26.0 ± 0.3 Å. In the elution profile of 1.5 M GdnHCl it was also possible to detect the unfolded state of trypsinogen, whose area increased to a single peak at 3 M GdnHCl. These results indicate the presence of a heterogeneous intermediate population in the unfolding process of trypsinogen.
- Size exclusion in HPLC of trypsinogen GdnHCl unfolding. The column (Shodex - Protein KW 803 (0.8 x 30 cm), Showa Denko & Shoko) was loaded with 50 µg of protein per run and equilibrated with 100 mM MES, pH 7.0, 20 mM CaCl2 and 81 mM NaNO3 buffer, pH 7.0, containing GdnHCl from 0 M to 3 M, respectively. Trypsinogen concentration was 1 mM in each sample. The flow rate was 1 ml/min and elution was monitored by absorbance at 280 nm. The profiles are shown with their respective retention times in minutes and the concentration of GdnHCl in molar concentration. The Stokes radii were calculated according to Uversky (21).
Equilibrium unfolding of trypsinogen induced by urea
Figure 3 presents the denaturation curve of trypsinogen at pH 7.0 and 25oC using urea as denaturant. The fU was plotted as a function of urea concentration. The curve was monitored by the differential spectra at 293 nm (DS) and circular dichroism ([Q]) at 300 nm. The non-coincidence of the curves measured by different techniques demonstrates the existence of one or more intermediates in the unfolding process. The denaturation curves were fitted to a two-state model. Table 1 shows the results of this analysis.
- Unfolding transition of trypsinogen in 100 mM MES, pH 7.0, 20 mM CaCl2 and 81 mM NaNO3, pH 7.0, as a function of urea concentration. The fraction fU was calculated from the molar ellipticity at 300 nm from circular dichroism (circles) and from difference spectroscopy signal at 293 nm (triangles). The near-UV CD was monitored at 300 nm using a 0.5-cm path length cuvette. The difference spectroscopic measurements were made in a 0.5-cm path length tandem cell as described in the procedures. Protein concentration was 1 mM for each sample and temperature 25oC. The dashed line represents a two-state fit of the CD 300-nm data calculated according to Santoro and Bolen (24). The solid line represents the two-state fit of DS 293-nm data. The statistical analysis of the data is presented in Table 1.
Size exclusion chromatography in urea and Stokes radius calculation
Figure 4 presents the urea-induced unfolding of trypsinogen at 25oC. SE-HPLC was used to determine the dimensions of the conformers and the Stokes radii calculated for native and unfolded states were in agreement with those reported in the literature (22.4 Å for native trypsinogen and 29.2 Å for unfolded state) (27). The RS of the native state was 19.8 ± 0.2 Å and for the unfolded state was 28.1 ± 0.2 Å. The data for the unfolding of trypsinogen in urea are similar to that observed for guanidine denaturation, i.e., the occurrence of an intermediate peak at 2 M urea with an RS of 23.9 ± 0.3 Å. This fraction changed in position during denaturation at 3 M with a corresponding RS of 25.7 ± 0.6 Å. A third peak was identified at 5 M urea with an RS of 25.9 ± 0.4 Å. The denaturation of trypsinogen can be presented as two successive denaturation transitions, the first separating the compact and slightly more swollen protein molecules, and the second corresponding to a further unfolding of this intermediate up to the completely unfolded state.
- Size exclusion in HPLC of trypsinogen urea denaturation. The column (Shodex - Protein KW 803 (0.8 x 30 cm), Showa Denko & Shoko) was loaded with 50 µg of protein per run and equilibrated with 100 mM MES, pH 7.0, 20 mM CaCl2 and 81 mM NaNO3, pH 7.0, containing urea at different concentrations. Trypsinogen concentration was 1 mM in each sample. The flow rate was 0.75 ml/min and elution was monitored by absorbance at 280 nm. Profiles present the retention times in minutes for each urea concentration. The Stokes radii were calculated according to Uversky (21).
ANS fluorescence on binding
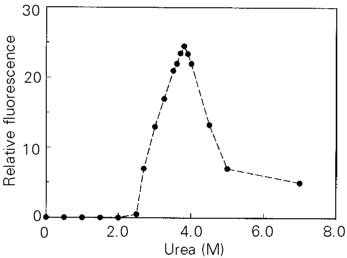
Usually the hydrophobic core of globular proteins is well protected from solvents by the rigid packing of the side chain organized in the tertiary structure. This feature results in low affinity for hydrophobic probes such as ANS. The hydrophobic side chains in fully unfolded proteins that practically have no secondary structure and are not compact also do not bind ANS. The affinity of ANS for the protein increases when the rigidity of the tertiary structure of the protein is disrupted, while the secondary structure and compactness are retained. The highest affinity of ANS for these conformers identifies the molten globule state (3). Figure 5 shows a significant increase in the fluorescence of the ANS probe binding to trypsinogen at around 3.8 M urea, indicating that at this denaturant concentration there should be a swollen compact intermediate with solvent-exposed hydrophobic areas, i.e., a molten globule.
- Urea dependence of ANS binding by bovine trypsinogen measured by intensity of ANS fluorescence at 480 nm (excitation at 350 nm). The samples were incubated for 15 min. The excitation and emission bandwidths were fixed at 2 nm each and a 1-cm path length quartz cuvette was used for all measurements. Protein concentrations were 500 µM for all assays and temperature was 25oC.
Results
The unfolding of trypsinogen induced by guanidine chloride, monitored in the tertiary structure, showed quite coincident data, suggesting a highly cooperative denaturation fitting a two-state model (no apparent intermediate) (Figure 1). On the other hand, following denaturation in the far-UV region, the transition showed less cooperativity and a quite complex behavior, fitting a three-state model (apparent intermediate states). These results suggest that the denaturation of trypsinogen consists of two successive phases - the first with loss of tertiary structure and exposure of hydrophobic residues reaching an intermediate state, followed by a second phase with loss of secondary structure reaching the completely unfolded state.
Additionally, the exposure of new hydrogen bonding groups and of hydrophobic groups during the denaturation usually increases the amount of water bound to the protein molecule. This feature has been detected by SE-HPLC, which revealed a second population of intermediates (Figure 2). The Stokes radius calculation showed an intermediate conformer with dimensions between those of the native and the unfolded forms of trypsinogen.
The urea unfolding of trypsinogen (Figure 3) showed non-coincidence transition in the near-UV circular dichroism and in difference spectral data. These results indicate the presence of an intermediate state of equilibrium and reflect the decrease of the near-UV CD data, in other words, the N  I transition. The unfolding of trypsinogen followed by DS at 293 nm was fitted to a two-state model, but did not coincide with the CD transition. The denaturation curves, taken together, describe two different phases, the first one with loss of tertiary structure followed by the hydrophobic exposure of tryptophan residues, indicating intermediates in this pathway.
I transition. The unfolding of trypsinogen followed by DS at 293 nm was fitted to a two-state model, but did not coincide with the CD transition. The denaturation curves, taken together, describe two different phases, the first one with loss of tertiary structure followed by the hydrophobic exposure of tryptophan residues, indicating intermediates in this pathway.
Furthermore, the intermediates detected by SE-HPLC showed Stokes radii varying from 24.1 Å to 26.0 Å in GdnHCl and from 23.9 Å to 25.9 Å in urea denaturation (Figures 2 and 4). Based on these data, the minimum unfolding model for bovine trypsinogen compatible with our overall experimental data should be: N I U. Indeed, the intermediate population proved to be heterogeneous.
The possible existence of a molten globule in the presence of about 4 M urea was reinforced by the binding of the fluorophobic probe 1,8 ANS, as seen in Figure 5. The binding of the fluorescent dye is considered to be an identification of the molten globule state based on the high affinity of this conformer to the probe. The affinity is due to the absence of rigid packing of hydrophobic clusters in this state and, hence, to a greater accessibility of the protein hydrophobic core for a solvent. The specificity of the binding is due to additional electrostatic interactions of ANS with the molecule of the molten globule; these interactions cannot occur in the native or in the completely unfolded state (22).
The present data suggest that the unfolding of trypsinogen follows a complex pathway with one or more intermediates. These results are in good agreement with the observation of an MG for pressure denatured trypsin (14) and for the kinetic intermediate in trypsinogen unfolding (11).
Discussion
Acknowledgments
The authors thank to Dr. Michel Goldberg and Dr. Alain Chaffotte, Institute Pasteur, Paris for helpful discussion.
Address for correspondence: M.M. Santoro, Departamento de Bioquímica e Imunologia, ICB, UFMG, Av. Antonio Carlos, 6627, Caixa Postal 486, 30161-970 Belo Horizonte, MG, Brasil. Fax: +55-31-441-5963. E-mail: martins@red.incm.u-nancy.fr and santoro@mono.icb.ufmg.br
Research supported by FAPEMIG and CAPES. N.F. Martins was the recipient of a fellowship from CAPES. Part of a Ph.D. thesis presented by N.F. Martins to the Departamento de Bioquímica e Imunologia, UFMG, Belo Horizonte, MG. Received June 17, 1998. Accepted February 26, 1999.
- 1. Ohgushi M & Wada A (1983). Molten globule state: a compact form of globular proteins with mobile side-chains. FEBS Letters, 164: 21-24.
- 2. Sagami M, Eras S, Koseki T & Nagai N (1998). Structural characterization of the molten globule and native states of ovalbumin: a 1 H NMR study. Journal of Peptide Research, 50: 465-474.
- 3. Semistonov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF & Gilmanshin RI (1991). Study of the molten globule intermediate state by a hydrophobic fluorescent probe. Biopolymers, 31: 119-128.
- 4. Ewbank JJ, Creighton TE, Hayer-Hartl MK & Ulrich Hartl F (1995). What is a molten globule? Structural Biology, 2: 10-11.
- 5. Dolgikh DA, Gilmanshin RI, Brazhnikov EV, Bychkova VE, Semistonov GV, Venyaminov SY & Ptitsyn OB (1981). a-Lactalbumin:compact state with fluctuating tertiary structure? FEBS Letters, 136: 311-315.
- 6. Wittung SP (1998). A stable molten-globule-like cytochrome C. Biochimica et Biophysica Acta, 1382: 324-332.
- 7. Chaffotte AF, Guijarro JI, Guillou Y, Delepierre M & Goldberg ME (1997). The "pre-molten globule", a new intermediate in protein folding. Journal of Protein Chemistry, 16: 433-439.
- 8. Uversky VN & Ptitsyn OB (1996). Further evidence on the equilibrium "pre-molten globule state" four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. Journal of Molecular Biology, 255: 215-228.
- 9. Haynie DT & Freire E (1993). Structural energetics of the molten globule state. Proteins, 16: 115-140.
- 10. Bode W, Fehlamer H & Huber R (1976). Crystal structure of bovine trypsinogen at 1.8 Ĺ resolution. Journal of Molecular Biology, 106: 325-335.
- 11. Bulaj G & Otlewski J (1994). Denaturation of free and complexed bovine trypsinogen with the calcium ion, dipeptide Ile-Val and basic pancreatic trypsinogen inhibitor (kunitz). European Journal of Biochemistry, 223: 939-946.
- 12. Bulaj G & Otlewski J (1995). Ligand-induced changes in the conformational stability of bovine trypsinogen and their implications for the protein function. Journal of Molecular Biology, 247: 701-706.
- 13. Otlewski J, Sywula A, Kolasinski M & Krowarsch D (1996). Unfolding kinetic of bovine trypsinogen. European Journal of Biochemistry, 242: 601-607.
- 14. Ruan K, Lange R, Bec N & Balny C (1997). A stable partly denatured state of trypsin induced by high hydrostatic pressure. Biochemical and Biophysical Research Communications, 239: 150-154.
- 15. Nozaki Y (1972). The preparation of guanidine hydrochloride. In: Hirs CHW & Timasheff SN (Editors), Methods in Enzymology Academic Press, New York, 43-50.
- 16. Ascenzi PL, Menegati E, Bortolotti F, Guarnieri M & Antonini E (1981). Steady state and pre steady state kinetics of the trypsin catalised hydrolisis of a-CBZ-L-lysine-p-nitrophenylester. Biochimica et Biophysica Acta, 658: 158-164.
- 17. Laemmli UK (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685.
- 18. Croft LR (1973). Amino and carboxy terminal sequence of crystallin from hadbock lens. Biochimica et Biophysica Acta, 295: 174-177.
- 19. Chang JY (1977). High sensitivity sequence analysis of peptides and proteins by 4-NN-dimethylaminoazobenzene 4-isothiocyanate. Biochemical Journal, 163: 517-520.
- 20. Johnson Jr WC (1990). Protein secondary structure and circular dichroism: a practical guide. Proteins, 7: 205-214.
- 21. Uversky VN (1993). Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry, 32: 13288-13298.
- 22. Uversky VN, Winters S & Lober G (1996). Use of fluorescence decay time of 8-ANS-protein complexes to study the conformational transitions in proteins which unfold through the molten globule state. Biophysical Chemistry, 60: 79-88.
- 23. Pace CN (1986). Determination and analysis of urea and guanidine hydrochloride denaturation curves. In: Hirs CHW & Timasheff SN (Editors), Methods in Enzymology Academic Press, New York, 266-280.
- 24. Santoro MM & Bolen DW (1992). A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry, 20: 4901-4907.
- 25. Matthews RC & Crisanti MM (1981). Urea induced unfolding of the a-subunit of tryptophan synthase evidence for a multistate process. Biochemistry, 20: 784-792.
- 26. Izquierdo C, Burguillo FJ, User JL & Del Arco A (1987). Kinetic study of the inhibition of amidolytic activity of thrombin by benzamidine and N-dansyl-(p-guanidine)-phenylalanine-peptide. International Journal of Biochemistry, 19: 1105-1112.
- 27. Light A & Higaki JN (1987). Detection of intermediate species in the refolding of bovine trypsinogen. Biochemistry, 26: 5556-5564.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
20 July 1999 -
Date of issue
June 1999
History
-
Accepted
26 Feb 1999 -
Received
17 June 1998