Abstract
The protease ZapA, secreted by Proteus mirabilis, has been considered to be a virulence factor of this opportunistic bacterium. The control of its expression requires the use of an appropriate methodology, which until now has not been developed. The present study focused on the replacement of azocasein with fluorogenic substrates, and on the definition of enzyme specificity. Eight fluorogenic substrates were tested, and the peptide Abz-Ala-Phe-Arg-Ser-Ala-Ala-Gln-EDDnp was found to be the most convenient for use as an operational substrate for ZapA. A single peptide bond (Arg-Ser) was cleaved with a Km of 4.6 µM, a k cat of 1.73 s-1, and a catalytic efficiency of 376 (mM s)-1. Another good substrate for ZapA was peptide 6 (Abz-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-Gln-EDDnp) which was cleaved at a single bond (Phe-Ser) with a Km of 13.6 µM, a k cat of 3.96 s-1 and a catalytic efficiency of 291 (mM s)-1. The properties of the amino acids flanking the scissile bonds were also evaluated, and no clear requirement for the amino acid residue at P1 was found, although the enzyme seems to have a preference for a hydrophobic residue at P2.
metalloprotease; substrate specificity; quenched fluorescence peptides; Proteus mirabilis
Braz J Med Biol Res, July 2000, Volume 33(7) 765-770
Development of an operational substrate for ZapA, a metalloprotease secreted by the bacterium Proteus mirabilis
B.L. Fernandes1, M.A.F. Anéas1, L. Juliano2, M.S. Palma3, I. Lebrun4 and F.C.V. Portaro4
1Departamento de Microbiologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, SP, Brasil
2Departamento de Biofísica e Biologia Molecular, Universidade Federal de São Paulo, São Paulo, SP, Brasil
3Departamento de Biologia, Instituto de Ciências Biológicas, Universidade Estadual de São Paulo, Rio Claro, SP, Brasil
4Departamento de Bioquímica e Biofísica, Instituto Butantan, São Paulo, SP, Brasil
References
Abstract
The protease ZapA, secreted by Proteus mirabilis, has been considered to be a virulence factor of this opportunistic bacterium. The control of its expression requires the use of an appropriate methodology, which until now has not been developed. The present study focused on the replacement of azocasein with fluorogenic substrates, and on the definition of enzyme specificity. Eight fluorogenic substrates were tested, and the peptide Abz-Ala-Phe-Arg-Ser-Ala-Ala-Gln-EDDnp was found to be the most convenient for use as an operational substrate for ZapA. A single peptide bond (Arg-Ser) was cleaved with a Km of 4.6 µM, a kcat of 1.73 s-1, and a catalytic efficiency of 376 (mM s)-1. Another good substrate for ZapA was peptide 6 (Abz-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-Gln-EDDnp) which was cleaved at a single bond (Phe-Ser) with a Km of 13.6 µM, a kcat of 3.96 s-1 and a catalytic efficiency of 291 (mM s)-1. The properties of the amino acids flanking the scissile bonds were also evaluated, and no clear requirement for the amino acid residue at P1 was found, although the enzyme seems to have a preference for a hydrophobic residue at P2.
Key words: metalloprotease, substrate specificity, quenched fluorescence peptides, Proteus mirabilis
Introduction
The proteins albumin, azocasein and denatured hemoglobin were extensively used in the past to detect and partially characterize new proteolytic enzymes (see Ref. 1 for a review). However, difficulties in monitoring enzymatic activity, the low sensitivity of these methods, and the comparison of the data obtained with the properties of other well-characterized enzymes were some of the problems when these proteins were used as substrates to define novel proteases. More importantly, these substrates were unsuitable for the determination of enzyme specificity, both concerning the most susceptible scissile bond cleaved by the enzyme and the requirements for specific amino acid side chains in the primary sequence of the substrate. Thus, the development of synthetic peptide substrates containing chromogenic or fluorogenic groups (1,2) greatly accelerated the progress of the methodologies used to isolate and characterize new proteolytic enzymes.
We have been studying the genetic instability of ZapA expression (3), a metalloprotease which is considered to be a virulence factor of Proteus mirabilis (4). In order to pursue the project concerning the genetic control of ZapA expression by P. mirabilis, an appropriate methodology for enzyme detection was required. Surprisingly, azocasein had been the only substrate employed (5). In this study we analyzed several quenched fluorogenic substrates to identify the peptide with the best kinetic properties to be used as an operational substrate in the further characterization of this enzyme and its role in P. mirabilis pathogenicity.
Material and Methods
Bacterial strain and growth condition
Escherichia coli strain DH5a [F-, f80dlacZDM15 D(lacZYA-argF) U169 endA1 recA1 hsdR17 (rK- mK+) deoR thi-1 supE44 l-gyrA 96 relA1] carries plasmid pCW101, which contains the whole zap gene cluster (5). LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl) supplemented with ampicillin (50 µg/ml) was used for E. coli growth.
Peptide substrates
The intramolecularly quenched fluorescent peptide substrates were synthesized, purified, and characterized by described procedures (2), using the multiple automated peptide synthesizer PSSM-8 from Shimadzu Corporation (Tokyo, Japan). These peptides contain an Abz-group (ortho-aminobenzoyl) at the N-terminus, and EDDnp (2,4-dinitrophenyl-ethylenediamine) at the C-terminus, as fluorescent and quencher pair, respectively. All peptides used in this study are listed in Table 1.
Protease purification by phenyl-Sepharose affinity chromatography
The recombinant ZapA protease was purified as described (5), with little modification. Briefly, the culture supernatant of overnight grown bacteria was centrifuged (7000 g, 30 min, 4oC) and filtered through 0.45-µm pore-size Millipore filters. The filtrates were loaded at a flow rate of 1 ml/min at 4oC onto columns (2 x 60 cm) of phenyl-Sepharose (Pharmacia, Uppsala, Sweden) equilibrated in 50 mM Tris-HCl, pH 8.0. Columns were then washed with 10 volumes of the same buffer. Bound protease was eluted with 50 mM Tris-HCl, pH 11.0, and the collected fractions (3.8 ml) were monitored for protein content with a spectrophotometer at A280 nm, after the pH had been adjusted to pH 8.0. The peak fractions were pooled, and concentrated by differential centrifugation using Millipore Ultrafree filters (size cut-off, 30 kDa). Material retained by the filter was aliquoted at appropriate concentrations and kept at -20oC in 15% glycerol. The homogeneity of the preparation was analyzed by SDS-PAGE (6).
Enzymatic assays
Hydrolysis of the intramolecularly fluorogenic quenched peptide substrates at 37oC in 50 mM Tris-HCl, 2 mM CaCl2, pH 8.0, was monitored by measuring fluorescence at lem = 420 nm and lex = 320 nm with a Hitachi F-2000 spectrofluorometer, as previously described (7). Standard hydrolysis conditions were strictly maintained for different substrates. The enzyme concentrations varied from 2.8 pM for the best substrates to 8.4 pM for the less susceptible ones. In most cases, substrate concentration ranged from 10 times lower than the Km to 10 times higher than the Km. The kinetic parameters were calculated according to Wilkinson (8). The fluorogenic peptide 1 was used to determine the activities of the recombinant enzyme. One unit of ZapA activity is the amount of enzyme which hydrolyzes 1 µmol of peptide 1 in 1 min. All enzyme assays were performed in triplicate.
HPLC analysis of synthetic fluorogenic substrates and of their enzymatic hydrolysis products
The peptide solutions (20-50 µM) in 50 mM Tris-HCl, pH 8.0, and 2 mM CaCl2 were incubated with the native and the recombinant proteases at 37oC. Samples (500 µl) from the substrate and reactions were periodically removed for HPLC analysis until 100% hydrolysis was achieved. The hydrolysis products were separated by HPLC, collected manually, and submitted to mass spectrometry. The scissile bonds were deduced from the sequences of the substrate fragments. The HPLC conditions used for the analytical procedure were 0.1% trifluoroacetic acid in water (solvent A) and acetonitrile solvent A (9:1) as solvent B. The separations were performed at a flow rate of 1 ml/min using a J.T. Baker C-18 column (4.6 x 300 mm). Analytical HPLC was performed using an SPD-10AV Shimadzu UV/visible detector, and an RF-10 AX fluorescence detector. For peptide purification, a semi-prep Ultrapac TSK C-18-120T column (7.8 x 300 mm, 10 µm particles; LKB, Bromma, Sweden) was used with the same solvents as above at a flow rate of 2 ml/min. In all cases, elution was followed by ultraviolet absorption (214 nm) and by fluorescence (lem = 420 nm and lex = 320 nm).
Mass spectrometry
The mass spectrometry experiments were performed using a QUATTRO II triple quadrupole mass spectrometer equipped with a standard ES probe (Micromass, Manchester, UK) adjusted to ca. 40 µl/min with an ODS-HG-5 microcolumn (0.3 mm ID x 150 mm, 5 µm particles), using a stream splitter at a ratio of approximately 1:15. The mass spectrometer data acquisition and treatment system was equipped with the MassLynx and MaxEnt software for handling spectra. During all experiments the source temperature was maintained at 80oC and the needle voltage at 3.6 kV, applying a drying gas flow (nitrogen) of 200 l/h and a nebulizer gas flow of 20 l/h. The mass spectrometer was calibrated with intact horse heart myoglobin and its typical cone voltage-induced fragments. For reliable mass determination of the intact ß-chain of insulin and its proteolytic fragments, reduction and carboxymethylation were performed as previously described (9). Mass spectrometric detection was achieved with different parameters for each type of experiment. The LC-separated peptides were detected by scanning from m/z 50 to 2000 at 6 s/scan, with a 31-V cone. Product ions from MS/MS experiments were detected in several runs by scanning the appropriate mass for each situation, using high energy (25 eV) for single charged precursor ions and low collision energy (15 eV) for multiple charged precursor ions. No tandem MS was recorded for peptides smaller than four amino acid residues.
Results and Discussion
A small series of fluorescent substrates used to characterize cathepsin B (10) proved to be adequate to determine the specificity of the metalloprotease secreted by Proteus mirabilis, ZapA. Peptide 1 (Abz-A-F-R-S-A-A-Q-EDDnp) showed the best catalytic efficiency (Table 1) and therefore was selected for use as an operational peptide substrate. It should replace the azocaseine method, introducing a series of obvious advantages in the studies involving this enzyme.
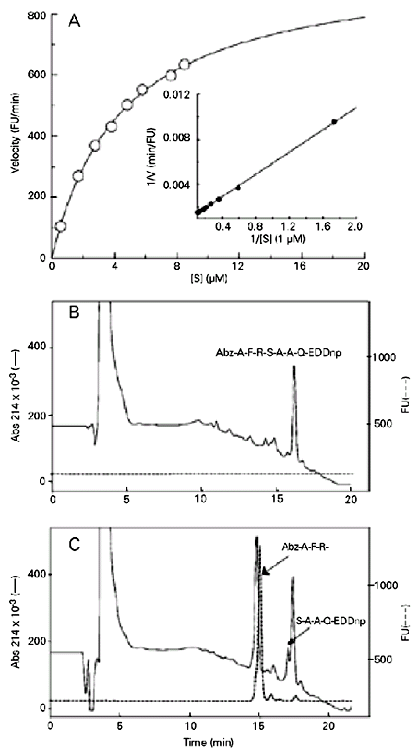
Figure 1 (panel A) shows the Michaelis-Menten and the Lineweaver-Burk plots (1/V x 1/[S] (11) obtained with peptide 1 hydrolyzed by ZapA. The other fluorogenic peptide substrates tested presented the same Michaelis-Menten profiles (data not shown). Panel B illustrates the HPLC elution profile of the intact peptide substrate 1, detected by fluorescence and absorbance at 214 nm. Panel C shows the HPLC elution profile of the two fragments generated by the hydrolytic activity of the ZapA metalloprotease on peptide 1, detected by absorbance at 214nm. Only the peak corresponding to the N-terminal fragment showed fluorescence, while the other fragment was only detected by UV absorbance. The peptide bond cleaved was Arg-Ser (see Table 1), as determined by mass spectrometry.
In the series of related substrates (peptides 1-5, Table 1) we found that, on the P' site (according to the nomenclature of Schechter and Berger, Ref. 12), the replacement of one alanine with aspartic acid (peptide 2), or the removal of one alanine (peptide 3) reduced the catalytic efficiency of ZapA by one order of magnitude, and the replacement of lysine at position P3 with aspartic acid further reduced the catalytic efficiency by one order of magnitude (peptide 4). In contrast, the introduction of a bulky hydrophobic residue, tryptophan, at position P4, significantly improved the binding, resulting in the lowest Km value in the series of peptides used in this study (peptide 5). For this whole series of substrates, a single bond, only the Arg-Ser bond, was cleaved.
For the other quenched fluorescent substrates used, displaying rather different amino acid sequences, we found that the bradykinin-derived peptide (peptide 6) represented the best substrate, displaying a kcat/Km value only about 23% lower than that found for peptide 1. The only peptide bond cleaved was the Phe-Ser bond. Another quenched fluorescent substrate, which has a hydrophobic residue at position P1, was the peptide whose sequence was derived from dynorphin (peptide 7), and was hydrolyzed at the Leu-Arg bond.
Finally, in peptide 8, whose amino acid sequence was derived from synaptobrevin (the natural substrate for the tetanus toxin metalloprotease (13)), the only peptide bond cleaved was the Glu-Thr bond, this turning out to be the worst substrate in the series of peptides used in this study.
The limited number of substrates studied does not permit a more detailed description of the enzyme specificity. However, it seems likely that a hydrophobic residue in P2 favors the catalytic efficiency of ZapA. The exception is peptide 6, which has Gly at position P2. This substrate has a good catalytic efficiency (kcat/Km = 291 (mM s)-1), but mostly due to the high kcat value. Thus, it seems likely that a hydrophobic residue in P2 is important to increase the affinity of the substrate for the enzyme. These findings agree with the substrate requirements of other bacterial metalloproteases, which also belong to the serralysin family of bacterial alkaline proteases (14).
The S1 site of ZapA showed broad specificity, efficiently accommodating substrates presenting an arginine, a phenylalanine, a leucine or a glutamic acid residue in P1 (see Table 1, peptides 1-8). The latter peptide offered an additional reason for hydrolysis at the Leu-Arg bond, which is the presence of Phe at position P2. In contrast, the results obtained with peptide 8 (Table 1) yielded the lowest value for kcat, indicating that an acidic residue at position P1 does not favor hydrolysis, even though a hydrophobic residue is located at position P2.
Investigations of the natural substrates of ZapA as well as the genetic control of its expression will be greatly facilitated by the method described here.
Address for correspondence: B.L. Fernandes, Departamento de Microbiologia, ICB II, USP, Av. Prof. Lineu Prestes, 1374, 05508-900 São Paulo, SP, Brasil. Fax: +55-11-818-7240.E-mail: blfernan@usp.br
Research supported by FAPESP and PIBIC/CNPq/USP. Received November 4, 1999. Accepted March 10, 2000.
- 1. Barrett AJ (1977). Proteinases in Mammalian Cells and Tissues Elsevier/North-Holland Biomedical Press, Amsterdam, The Netherlands.
- 2. Hirata IY, Cezari MHS, Nakaie CR, Boschcov P, Ito AS, Juliano MA & Juliano L (1994). Internally quenched fluorogenic protease substrates: solid-phase synthesis and fluorescence spectroscopy of peptides containing ortho-aminobenzoyl/dinitrophenyl groups as donor-acceptor pairs. Letters in Peptide Science, 1: 299-308.
- 3. Costa SOP (1990). A review of protease secretion instability in Proteus mirabilis Brazilian Journal of Genetics, 13: 165-172.
- 4. Rozalski A, Sidorczyk Z & Kotelko K (1997). Potential virulence factors of Proteus bacilli. Microbiology and Molecular Biology Reviews, 61: 65-89.
- 5. Wassif C, Cheek D & Belas R (1995). Molecular analysis of a metalloprotease from Proteus mirabilis Journal of Bacteriology, 177: 5790-5798.
- 6. Laemmli UK (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685.
- 7. Chagas JR, Juliano L & Prado ES (1991). Intramolecularly quenched fluorogenic tetrapeptide substrates for tissue and plasma kallikreins. Analytical Biochemistry, 192: 419-425.
- 8. Wilkinson GN (1961). Statistical estimations in enzyme kinetics. Biochemical Journal, 80: 324-332.
- 9. Van Baar BLM, Hulst AG & Wils ER (1999). Characterization of cholera toxin by liquid chromatography - electrospray mass spectrometry. Toxicon, 37: 85-98.
- 10. Portaro FCV, Santos ABF, Juliano MA, Cezari MHA, Juliano L & Carmona E (2000). Probing the specificity of cysteine proteinases at subsites remote from the active site: analysis of P4, P3, P2' and P3' variations in extended substrates. Biochemical Journal, 347: 123-129.
- 11. Segel IM (1975). Enzyme Kinetics John Wiley and Sons, New York.
- 12. Schechter I & Berger A (1967). On size of the active site in proteases. Biochemical and Biophysical Research Communications, 27: 157-162.
- 13. Yamazaki S, Baumeister A, Binz T, Blasi J, Link E, Cornille F, Roques B, Fykse EM, Sudhof TC, Jahn R & Niemann H (1994). Cleavage of members of the synaptobrevin VAMP family by type E and F botulinical neurotoxin and tetanus toxin. Journal of Biological Chemistry, 269: 12764-12772.
- 14. Maeda H & Morihara K (1995). Serralysin and related bacterial proteinases. Methods in Enzymology, 248: 395-413.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
14 June 2000 -
Date of issue
July 2000
History
-
Accepted
10 Mar 2000 -
Received
04 Nov 1999