
Abstract
Hydrolysis of D-valyl-L-leucyl-L-arginine p-nitroanilide (7.5-90.0 µM) by human tissue kallikrein (hK1) (4.58-5.27 nM) at pH 9.0 and 37ºC was studied in the absence and in the presence of increasing concentrations of 4-aminobenzamidine (96-576 µM), benzamidine (1.27-7.62 mM), 4-nitroaniline (16.5-66 µM) and aniline (20-50 mM). The kinetic parameters determined in the absence of inhibitors were: Km = 12.0 ± 0.8 µM and k cat = 48.4 ± 1.0 min-1. The data indicate that the inhibition of hK1 by 4-aminobenzamidine and benzamidine is linear competitive, while the inhibition by 4-nitroaniline and aniline is linear mixed, with the inhibitor being able to bind both to the free enzyme with a dissociation constant Ki yielding an EI complex, and to the ES complex with a dissociation constant Ki', yielding an ESI complex. The calculated Ki values for 4-aminobenzamidine, benzamidine, 4-nitroaniline and aniline were 146 ± 10, 1,098 ± 91, 38.6 ± 5.2 and 37,340 ± 5,400 µM, respectively. The calculated Ki' values for 4-nitroaniline and aniline were 289.3 ± 92.8 and 310,500 ± 38,600 µM, respectively. The fact that Ki'>Ki indicates that 4-nitroaniline and aniline bind to a second binding site in the enzyme with lower affinity than they bind to the active site. The data about the inhibition of hK1 by 4-aminobenzamidine and benzamidine help to explain previous observations that esters, anilides or chloromethyl ketone derivatives of Nalpha-substituted arginine are more sensitive substrates or inhibitors of hK1 than the corresponding lysine compounds.
kinetics of human tissue kallikrein inhibition; tissue kallikrein; 4-nitroaniline; aniline; benzamidine; 4-aminobenzamidine; enzyme inhibition
Braz J Med Biol Res, January 2001, Volume 34(1) 35-44
Linear competitive inhibition of human tissue kallikrein by 4-aminobenzamidine and benzamidine and linear mixed inhibition by 4-nitroaniline and aniline
M.O. Sousa1, T.L.S. Miranda2, E.B. Costa1, E.R. Bittar3, M.M. Santoro3 and A.F.S. Figueiredo1
Departamentos de 1Análises Clínicas e Toxicológicas, Faculdade de Farmácia, 2Engenharia Química, Escola de Engenharia, and 3Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brasil
References
Abstract
Hydrolysis of D-valyl-L-leucyl-L-arginine p-nitroanilide (7.5-90.0 µM) by human tissue kallikrein (hK1) (4.58-5.27 nM) at pH 9.0 and 37ºC was studied in the absence and in the presence of increasing concentrations of 4-aminobenzamidine (96-576 µM), benzamidine (1.27-7.62 mM), 4-nitroaniline (16.5-66 µM) and aniline (20-50 mM). The kinetic parameters determined in the absence of inhibitors were: Km = 12.0 ± 0.8 µM and kcat = 48.4 ± 1.0 min-1. The data indicate that the inhibition of hK1 by 4-aminobenzamidine and benzamidine is linear competitive, while the inhibition by 4-nitroaniline and aniline is linear mixed, with the inhibitor being able to bind both to the free enzyme with a dissociation constant Ki yielding an EI complex, and to the ES complex with a dissociation constant Ki', yielding an ESI complex. The calculated Ki values for 4-aminobenzamidine, benzamidine, 4-nitroaniline and aniline were 146 ± 10, 1,098 ± 91, 38.6 ± 5.2 and 37,340 ± 5,400 µM, respectively. The calculated Ki' values for 4-nitroaniline and aniline were 289.3 ± 92.8 and 310,500 ± 38,600 µM, respectively. The fact that Ki'>Ki indicates that 4-nitroaniline and aniline bind to a second binding site in the enzyme with lower affinity than they bind to the active site. The data about the inhibition of hK1 by 4-aminobenzamidine and benzamidine help to explain previous observations that esters, anilides or chloromethyl ketone derivatives of Na-substituted arginine are more sensitive substrates or inhibitors of hK1 than the corresponding lysine compounds.
Key words: kinetics of human tissue kallikrein inhibition, tissue kallikrein, 4-nitroaniline, aniline, benzamidine, 4-aminobenzamidine, enzyme inhibition
Introduction
The kallikreins (EC 3.4.21.8) are serine proteases present in glandular cells, neutrophils and biological fluids. They are divided into two main groups, i.e., plasma (EC 3.4.21.34) and tissue (EC 3.4.21.35) kallikreins. The two groups differ in molecular weight, isoelectric point, substrate specificity, immunological characteristics, the type of kinin released, and functional importance (1). Tissue kallikreins are related to trypsin, but with a higher specificity for the cleavage site of polypeptide substrates (2). Their principal known biological function is the highly selective hydrolysis of plasma high and low molecular mass kininogens at two different peptide bonds (Met379-Lys380 and Arg389-Ser390), with residues numbered on the basis of the structure of prekallikrein (3), to stoichiometrically release the vasoactive and spasmogenic decapeptide kallidin (Lys-bradykinin) (2). Human tissue kallikrein (hK1) (4) also hydrolyzes various synthetic substrates such as Na-substituted arginine and lysine derivatives (amides, esters and fluorogenic peptides) (5-8), Cbz-Tyr-OpNP (9,10) and D-Pro-Phe-Phe-Nan (8). Like other serine proteinases, hK1 is inhibited by diisopropylfluorophosphate, chloromethyl ketones of arginine and lysine, and basic pancreatic trypsin inhibitor (BPTI) (also known as aprotinin, Trasylol® or Kunitz pancreatic trypsin inhibitor) (6,11). In contrast, soybean trypsin inhibitor, a potent inhibitor of trypsin, plasma kallikrein and other serine proteinases, does not inhibit hK1 (6).
The inhibition of hK1 by BPTI is not a simple competitive inhibition as first reported (6,10), but is competitive inhibition of the parabolic type, with two inhibitor molecules binding to one enzyme molecule, forming a ternary enzymatic complex (12). It is noteworthy that the second BPTI molecule binds to the enzyme with higher affinity, suggesting that this second binding site was probably created or positively modulated as a consequence of the binding of the first BPTI molecule (12). However, the nature of the second binding site for this inhibitor in hK1 is still unknown (12).
There are controversial reports in the literature about the inhibition of hK1 esterase activity by benzamidine (BzA). BzA has been reported to be a competitive inhibitor of the hK1-catalyzed hydrolysis of Tos-Arg-OMe with a Ki value of 6.42 mM, but does not inhibit the hK1-catalyzed hydrolysis of Cbz-Tyr-OpNP even at a level of 2 mM (9). On the other hand, BzA has been reported to be a competitive inhibitor of the hK1-catalyzed hydrolysis of Cbz-Tyr-OpNP with a Ki value of 0.2 mM (10). However, there are no reports concerning the inhibition of the amidase activity of hK1 by 4-aminobenzamidine (4-ABzA), BzA, aniline (An) and 4-nitroaniline (4-NAn).
The purpose of the present study was to examine in depth the kinetics of the inhibition of hK1 amidase activity by 4-ABzA, BzA, 4-NAn and An in order to identify the precise mechanism of inhibition and to determine the number of binding sites and their accurate inhibition constants (Ki).
Our results clearly demonstrate that inhibition of hK1 amidase activity by 4-ABzA and BzA is a linear competitive inhibition with only one inhibitor molecule binding to one enzyme molecule, forming a binary enzymatic complex EI. On the other hand, our results also demonstrate that inhibition of hK1 amidase activity by An and 4-NAn is of the linear mixed type, with the inhibitors being able to bind both to the free enzyme to yield a binary enzymatic complex EI, and to the ES complex to yield a non-reactive ternary enzymatic complex ESI.
Material and Methods
Material
An, 4-NAn, 4-ABzA·2HCl, BzA·HCl and BPTI were purchased from Sigma Chemical Co. (St. Louis, MO, USA). D-Valyl-L-leucyl-L-arginine 4-nitroanilide (D-Val-Leu-Arg-Nan) was obtained from Chromogenix AB, (Mölndal, Sweden).
All other reagents were of reagent grade from Sigma.
Enzyme
Homogeneous hK1 was purified in our laboratory from healthy male urine (12). BPTI-titrated hK1 was used in the enzymatic assays (12).
Enzyme assay
Kallikrein amidase activity was assayed spectrophotometrically (6) at 410 nm to monitor the release of 4-NAn (e410 = 8800 M-1 cm-1). A 1-cm path-length cuvette containing 50 µl of 45.8-52.7 nM hK1 (Mr 31,000) (12) in 200 mM glycine/NaOH buffer, pH 9.0, containing 0.05% (w/v) NaN3 and 100 µl of buffer or 100 µl of an adequate dilution of a stock inhibitor solution (10 mM 4-ABzA, 100 mM BzA, 1.66 mM 4-NAn and 100-300 mM An) in the same buffer was placed in the thermostated cell compartment of a Shimadzu UV 160 A spectrophotometer at 37oC and pre-incubated for 5 min. The concentrations of 4-ABzA (e292= 15,280 M-1 cm-1) (13), BzA (e225 = 9390 M-1 cm-1) (13) and 4-NAn solutions were determined spectrophotometrically. Recently distilled aniline (d = 1.022 g/cm3) was used. The ranges of inhibitor concentrations were selected in order to obtain 20-80% inhibition. Then, 350 µl (10.7-128.6 µM) of D-Val-Leu-Arg-Nan in 200 mM glycine/NaOH buffer, pH 9.0, containing 0.05% (w/v) NaN3 was added, and the increase in absorbance at 410 nm with time was continuously recorded for 3 min. The slope of the time-dependent absorbance curve extrapolated to zero time was converted to µM of released 4-NAn per minute.
Bovine ß-trypsin (Bß-TR) (2.91 nM) was also assayed spectrophotometrically at 410 nm with the substrate D-Val-Leu-Arg-Nan (200-600 µM) in 100 mM Tris-HCl buffer, pH 8.1, containing 20 mM CaCl2 and 0.05% (w/v) NaN3, in the absence and in the presence of An (10-40 mM) in order to check for inhibition. Total substrate concentration was determined from the amount of 4-NAn released after complete hydrolysis by excess Bß-TR. In the inhibition assays with 4-NAn the reference cuvette contained the same amount of 4-NAn as the sample cuvette.
Treatment of kinetic data
The kinetic data of the inhibition of hK1 by 4-ABzA and BzA and the kinetic data of Bß-TR inhibition by An can be described, according to Plowman (14), as competitive inhibition.
On the other hand, the kinetic data of the inhibition of hK1 by 4-NAn and An can be described by the following scheme:

which, according to Cornish-Bowden (15), is the simplest formal mechanism for mixed inhibition. According to the author, "the inhibitor (I) can bind both to the free enzyme (E) to give a complex EI with dissociation constant Ki, and to the ES complex to give an unreactive ESI complex with dissociation constant Ki'. As shown in this scheme, both inhibitor-binding reactions are dead-end reactions and are therefore equilibria. As both EI and ESI exist, however, it is difficult to see why S should not bind directly to EI to give ESI. If this reaction is included in the mechanism the rate equation becomes much more complicated, because terms in S2 and I2 appear in it. These terms cancel only if all of the binding reactions are equilibria, i.e., if the substrate- and product-release steps are all fast compared with the reaction that converts ES into products. In practice, however, the predicted deviations from simple kinetics are difficult to detect experimentally, and one cannot use adherence to simple kinetics as evidence that Km, Ki and Ki' are true dissociation constants".
The normalized initial rate (v) will be given by the following equation:
According to Cornish-Bowden (15), when linear mixed inhibition occurs, both kcatapp, and kcatapp/Km app vary with the inhibitor concentration according to the following equations:
which can be rearranged to
Additionally,
and
which can be rearranged to
The kinetic parameters (Km, Kmapp, kcat, kcatapp and Ki for 4-ABzA, BzA, 4-NAn and An) were calculated based on unweighted non-linear regression analysis of the data fit to the appropriate Michaelis-Menten equations, while the Ki' values for 4-NAn and An were determined by fitting the experimental data to Equation 1. Whenever used, the linear replots (Equations 3 and 6) were shown only for diagnostic purposes; they were not used to calculate the Ki and Ki' values.

The hK1-catalyzed hydrolysis of D-Val-Leu-Arg-Nan followed Michaelis-Menten kinetics under the assayed substrate concentration range (7.5-90.0 µM). Figure 1 shows the 1/v vs 1/[S] plot for the hydrolysis of D-Val-Leu-Arg-Nan (7.5-90.0 µM) catalyzed by hK1 (4.58 nM) in the absence and in the presence of 4-ABzA (1.27-7.62 mM). The inset shows the replot of the slopes of the lines from Figure 1 (Kmapp/kcat appvs 4-ABzA concentration) according to Plowman (14). Each point is the mean of 4 determinations. Similar results were obtained with BzA.
Figure 2 shows the 1/v vs 1/[S] plot for the hydrolysis of D-Val-Leu-Arg-Nan (7.5-90.0 µM) catalyzed by hK1 (5.27 nM) in the absence and in the presence of 4-NAn (16.5-66 µM). Each point is the mean of 4 determinations. Similar results were obtained with An.
Figure 3 shows the replots of Kmapp/kcat app (panel A) and 1/kcatapp (panel B) vs 4-NAn concentration according to Cornish-Bowden (15) (Equations 6 and 3, respectively). The straight lines obtained are consistent with linear mixed inhibition. Statistical analysis of these data using the GraphPad program at the 95% confidence level showed that both lines have slopes that are significantly different from zero, and the statistical test for departure from linearity gave a negative (nonsignificant) result. Similar results were obtained with An. The kinetic parameters for hK1-catalyzed hydrolysis of D-Val-Leu-Arg-Nan in the absence and in the presence of 4-ABzA, BzA, 4-NAn and An are shown in Table 1.
The Bß-TR-catalyzed hydrolysis of D-Val-Leu-Arg-Nan followed Michaelis-Menten kinetics in the substrate concentration range assayed (200-600 µM) (data not shown).
The Michaelis-Menten plot for the hydrolysis of D-Val-Leu-Arg-Nan (200-600 µM) catalyzed by Bß-TR (2.91 nM) in the absence and in the presence of An (10-40 mM) showed enzyme inhibition (data not shown). The double-reciprocal plot for the An data showed convergent lines crossing at the same point on the 1/v axis, indicating competitive inhibition (data not shown). The replot of the slopes of the lines obtained from the double-reciprocal plot (Kmapp/kcat app) vs An concentration according to Plowman (14) showed a straight line with r2 = 0.952 (data not shown), indicating that An is a linear competitive inhibitor of Bß-TR amidase activity in the concentration range tested. No hint of a possible second binding site for An can be discerned in the experimental data obtained. The kinetic parameters (Km, kcat, kcat/Km and Ki) for the Bß-TR-catalyzed hydrolysis of D-Val-Leu-Arg-Nan in the absence and in the presence of An, determined according to Plowman (14), were 191.7 ± 38.2 µM, 1604 ± 107 min-1, 8.4 ± 1.8 min-1 µM-1 and 10.8 ± 1.0 mM, respectively.
- Lineweaver-Burk plot for the hydrolysis of D-Val-Leu-Arg-Nan by hK1 in the absence and in the presence of 4-ABzA. Inset, Kmapp/kcat appvs [4-ABzA]. Experimental conditions: 200 mM glycine/NaOH, pH 9.0, 37oC, 5-min incubation. hK1 concentration: 4.58 nM. 4-ABzA concentrations: closed circles, 0 mM; open circles, 0.096 mM; closed triangles, 0.192 mM; open triangles, 0.384 mM, and closed squares, 0.576 mM. Each point in the plot is the mean of quadruplicate determinations. More details are described in .
- Lineweaver-Burk plot for the hydrolysis of D-Val-Leu-Arg-Nan by hK1 in the absence and in the presence of 4-NAn. Experimental conditions: 200 mM glycine/NaOH, pH 9.0, 37oC, 3-min incubation. hK1 concentration: 4.58 nM. 4-NAn concentrations: closed circles, 0 µM; open circles, 16.5 µM; closed triangles, 33.0 µM; open triangles, 50.0 µM, and closed squares, 66.0 µM. Each point in the plot is the mean of quadruplicate determinations. More details are described in .
- Replots of Kmapp/kcat app (panel A) and of 1/kcatapp (panel B) against 4-nitroaniline concentration. Regression coefficients: for panel A - intercept: 0.29000 ± 0.01405; slope: 0.006152 ± 0.000347; r2: 0.9906; for panel B - intercept: 0.01898 ± 0.00057; slope: 0.00009593 ± 0.00001406; r2: 0.9395. More details are described in .
Results
During the characterization of our hK1 preparation, we decided to check the inhibition of its amidase activity by 4-ABzA (pKa2 = pKa of the amidinium group = 12.39), BzA (pKa = 11.41), 4-NAn (pKa = 1.0) (16) and An (pKa = 4.60) (16). At pH 9.0, the optimum pH for the hK1-catalyzed hydrolysis of D-Val-Leu-Arg-Nan, 4-ABzA and BzA showed a positive charge, while 4-NAn and An had no apparent electric charge, but dipole moments (µ) of 6.10 D and 1.53 D (16), respectively. 4-ABzA and BzA were chosen because they are good models of the side chains of arginine and lysine (17) in the usual human tissue kallikrein substrates (6,7). Aniline was also chosen because of its structural relationship to the side chain of the tyrosine constituent of the synthetic substrate CBz-Tyr-OpNP, which was demonstrated to be the hK1 substrate (9,10). On the other hand, 4-NAn was chosen not only because of its structural relationship to An, but also because it shows a larger dipole moment than the dipole moment of An (16); it is also one of the products of the hydrolysis of the chosen substrate.
hK1 inhibition by 4-ABzA and BzA
The double-reciprocal plot indicated that the hK1-catalyzed hydrolysis of D-Val-Leu-Arg-Nan is competitively inhibited by 4-ABzA (Figure 1). According to Plowman (14), competitive inhibition can be linear, hyperbolic, or parabolic. Thus, in order to distinguish the various types of competitive inhibition, it is necessary to replot the slopes of the lines obtained from the double-reciprocal plot vs inhibitor concentration. The result will be linear, hyperbolic or parabolic curves, distinguishing the various types of inhibition referred to as linear, hyperbolic or parabolic competitive inhibition, respectively. In order to clarify the type of inhibition of hK1 by 4-ABzA, it was decided to replot the slopes of the lines obtained from the double-reciprocal plot (Figure 1) vs 4-ABzA concentration. The results obtained showed a linear curve (Figure 1, inset), indicating that 4-ABzA is a linear competitive inhibitor (14) of hK1 amidase activity in the concentration range tested. Similar results were obtained with BzA (data not shown). The data in Table 1 are consistent with competitive inhibition since in the presence of 4-ABzA and BzA the Kmapp values increase with inhibitor concentration, although kcat values remain approximately constant (15). The present results are consistent with the model in which hK1 and 4-ABzA or BzA can form enzyme-inhibitor complexes with a stoichiometry of 1:1. No hint of a possible second binding site for these inhibitors can be discerned in the experimental data obtained. Similar results were reported for the 4-ABzA and BzA inhibitions of the amidase activity of trypsin, a well-known serine proteinase, which showed that 4-ABzA and BzA are potent competitive inhibitors with Ki values of 8.25 µM and 18.4 µM, respectively (17).
Comparison of the Ki values for 4-ABzA (pKa2 = 12.39) (146 ± 10 µM) and BzA (pKa = 11.41) (1,098 ± 91 µM) reveals that 4-ABzA is a 7.5-fold more potent hK1 inhibitor than BzA.
Comparison of the Ki values for hK1 inhibition by 4-ABzA (146 ± 10 µM) and BzA (1098 ± 91 µM) with the Ki for trypsin inhibition by 4-ABzA (8.25 µM) and BzA (18.4 µM) (17), respectively, reveals that 4-ABzA and BzA bind much more weakly to hK1 than to trypsin. These results agree with a published report stating that BzA binds much more weakly to hK1 (Ki ~15 mM) and pK1 (Ki ~1 mM) than to trypsin (Ki = 20 µM) (18).
The present results can explain previous observations regarding the hK1 substrate specificity, which indicate that arginine ester or anilide derivatives are more sensitive substrates for the enzyme than the corresponding lysine compounds (19). Similarly, our data explain previous observations about the reactivity of arginine and lysine chloromethyl ketones in inactivating hK1 which revealed that the enzyme was 10-fold more reactive with the Arg chloromethyl ketones than with the Lys ones (11). Thus, since the pKa value of the guanidinium group localized on the side chain of Arg is 12.5 and the pKa value of the amino group localized on the side chain of Lys is 10.0, it is easy to explain why hK1 has a significant preference for Arg over Lys residues at the P1 position (20) of their substrates (21). As previously reported, the stability of ion pairs increases with the difference in pKa of the groups involved (22). In this way, ion pairs formed by a given anion (for instance, carboxylate) with Arg will be more stable than those formed by Lys (22). There is evidence that this is true in many circumstances of biological interests (23,24). The interactions between hK1 and 4-ABzA and BzA seem to follow the same rule.
Comparison of the 4-ABzA and BzA inhibition of the amidase activities of human tissue kallikrein (this work) and trypsin (17) reveals that with these small molecules the inhibition mechanism of these two serine proteinases is similar - both enzymes are inhibited by a linear competitive mechanism.
hK1 inhibition by 4-NAn and An
The Michaelis-Menten plot for the hydrolysis of D-Val-Leu-Arg-Nan (7.5-90 µM) catalyzed by hK1 (4.58-5.27 nM) in the absence and in the presence of 4-NAn (pKa = 1.00) (16.5-66 µM) and An (pKa = 4.60) (20-50 mM) showed enzyme inhibition (data not shown). The double-reciprocal plot for the 4-NAn data (Figure 2) showed convergent lines crossing approximately at the same point in the second quadrant, indicating linear mixed inhibition (15). Similar results were obtained with An. The data for 4-NAn and An were also analyzed by the Dixon plot (1/v vs [I]) and by the Cornish-Bowden plot ([S]/v vs[I]) (15), respectively. The straight lines obtained from the Dixon plots intersected approximately at the same point in the second quadrant, while the straight lines obtained from the Cornish-Bowden plots intersected approximately at the same point in the third quadrant (data not shown). These results also indicate linear mixed inhibition (15). According to Segel (25), the Dixon plots for partial and most mixed-type inhibition systems are curved. However, when the ESI complex is not catalytically active, the plot is linear. The data for 4-NAn and An in Table 1 are also not consistent with competitive inhibition. As both Kmapp and kcatapp vary with inhibitor concentration, linear mixed inhibition is suggested (15).
Thus, in order to further clarify the inhibition type, we decided to replot the values of Kmapp/kcat app and 1/kcatappvs [I], respectively, according to Equations 6 and 3, respectively (Figure 3, panels A and B, shows the results obtained with 4-NAn). Similar results were obtained with An. As both Kmapp and kcatapp vary with [I], linear mixed inhibition was indicated (15). The Ki values for 4-NAn (38.6 ± 5.2 µM) and for An (37,340 ± 5,400 µM) and the Ki' values for 4-NAn (289.3 ± 92.8 µM) and for An (310,500 ± 38,600 µM), respectively, were calculated according to the fit of Equation 1 into the corresponding data in a Michaelis-Menten plot.
Comparison of the Ki values for hK1 inhibition by 4-NAn (pKa = 1.00) (38.6 ± 5.2 µM) and 4-ABzA (pKa2 = 12.39) (146 ± 10 µM), respectively, reveals that 4-NAn binds 3.8-fold more strongly to the active center of hK1 than 4-ABzA. Since at pH 9.0 the amidinium group of 4-ABzA is bearing a full positive charge, while the amino group of 4-NAn shows a positive charge induced by intramolecular transfer of electrons from the amino group to the nitro group by isovalent resonance (16,26), it would be reasonable to expect that 4-ABzA would interact better with the S1 subsite of the active center of hK1 (20) than 4-NAn. However, the data obtained do not concur with this reasoning. As a speculation, we may assume that 4-NAn, after binding to the S1 subsite of the active center of hK1 (20), possibly could participate in an additional enzyme-inhibitor interaction of the dipole-dipole type, involving the negative charge of the oxygen atom of the nitro group at the C-4 position of the aromatic ring, induced by resonance (16), with some group in the neighborhood of the active center region of hK1, while 4-ABzA, which shows an uncharged-NH2 group, also at the C-4 position of the aromatic ring, would not participate. The additional dipole-dipole interaction would reinforce the binding of 4-NAn to the active center of hK1.
A similar interaction was suggested by Mares-Guia et al. (26) to explain their results about the electronic effects in the interaction of para-substituted benzamidines with trypsin. According to these authors, their data can be interpreted in terms of an enzyme-inhibitor interaction of the dipole-dipole type. A dipole will appear in the inhibitor as a consequence of an intramolecular charge transfer from the substituent to the ring or vice-versa. Their data concur with the model in which intramolecular charge transfer renders positive the electron-donating substituent, thereby giving origin to a dipole that is able to interact with a site in the enzyme. According to the authors, the hydroxyl group of the reactive Ser183 is the most probable candidate for the dipole of the enzyme that interacts with the dipole at the para position in substituted benzamidines. On the other hand, electron-withdrawing groups generate a dipole of opposite polarity that decreases binding by a dipole-dipole repulsion.
We do not know which group is a possible candidate for the dipole of the enzyme that interacts with the dipole in 4-NAn.
Comparison of the Ki values for 4-NAn (38.6 ± 5.2 µM) and for An (37,340 ± 5,400 µM) reveals that 4-NAn binds 967-fold more strongly to the active center of hK1 than An. This result is partially consistent with the larger dipole moment of 4-NAn (6.10 D) over An (1.53 D) (16). However, the larger dipole moment of 4-NAn over An is not sufficient to explain the data obtained. As a speculation, we may state that, after binding to the S1 subsite of the active center of hK1 (20) through its =NH2+ group, An could not participate in an additional dipole-dipole interaction as 4-NAn can.
Comparison of the Ki' values for 4-NAn (289.3 ± 92.8 µM) and for An (310,500 ± 38,600 µM) reveals that 4-NAn binds 1073-fold more strongly to a second binding site on hK1 than An. The second binding site for these molecules is not known, but it is quite probable that the negative charge of the oxygen atom of the nitro group of the 4-NAn molecule is more available for an additional dipole-dipole interaction with it than the negative charge at the C-4 position of the An molecule.
The fact that Ki'>Ki indicates that both 4-NAn and An bind to a second binding site in the hK1 molecule with lower affinity than they bind to the S1 subsite of the hK1 active center.
The presence of a second BPTI binding site in hK1 has been already demonstrated (12). We do not know the location of this second binding site for 4-NAn or An in hK1, and also whether it is different from the second binding site for BPTI.
4-NAn is a special case since it is present in the substrate molecule D-Val-Leu-Arg-Nan where it shows neither a positive nor a negative charge. In the ES complex, the Nan (4-NAn) group is accommodated at the S1' position (20) of the hK1 active center. After substrate hydrolysis the released 4-NAn becomes a dipolar molecule. As a dipolar molecule 4-NAn is able to bind to the anionic site of hK1 as a competitive inhibitor through the positive charge on the amino group, and is also able to bind to a second binding site in hK1, possibly through the negative charge of the oxygen atom of the nitro group. Thus, 4-NAn is a product of the reaction that is able to inhibit hK1 as a mixed inhibitor.
The presence of a second inhibitor binding site in hK1 seems to be clear and may have important implications in the physiological activity of this enzyme.
Bß-TR inhibition by An
Comparison of the kcat/Km values for the hK1- and Bß-TR-catalyzed hydrolysis of D-Val-Leu-Arg-Nan (4.03 ± 0.43 and 8.4 ± 1.8 min-1 µM-1, respectively) reveals that D-Val-Leu-Arg-Nan is a slightly better substrate for Bß-TR than for hK1.
Additionally, comparison of the Ki values for An inhibition of hK1 (37,340 ± 5,400 µM) and of Bß-TR (10,800 ± 1,000 µM) reveals that An interacts better with the S1 subsite of the active center of Bß-TR than with the S1 subsite of the active center of hK1.
Discussion
Acknowledgments
We thank Prof. Giovanni Gazzinelli for reading the manuscript and offering constructive criticism.
Address for correspondence: A.F.S. Figueiredo, Departamento de Análises Clínicas e Toxicológicas, Faculdade de Farmácia, UFMG, Caixa Postal 689, 30123-970 Belo Horizonte, MG, Brasil. Fax: +55-31-339-7666. E-mail: afsf@farmacia.ufmg.br
Presented at the XXVIII Annual Meeting of the Brazilian Society of Biochemistry and Molecular Biology, Caxambu, MG, Brazil, May 22-25, 1999. This work is part of a PhD thesis to be presented by M.O. Sousa to the Graduate course in Biochemistry and Immunology, ICB, UFMG. Research supported by FAPEMIG (No. CBS 1173/95) and CNPq (No. 521259/95-9). T.S. Miranda was the recipient of a Recent Doctor fellowship from CNPq (No. 300716/95-8). E.B. Costa was the recipient of a Scientific Initiation fellowship from CNPq (No. 521259/95-9). E.R. Bittar is the recipient of a Graduate Student fellowship from CNPq. Received November 26, 1999. Accepted October 11, 2000.
- 1. Bhoola KD, Figueroa CD & Worthy K (1992). Bioregulation of kinins: kallikreins, kininogens and kininases. Pharmacological Reviews, 44: 1-80.
- 2. MacDonald RJ, Margolius HS & Erdös EG (1988). Molecular biology of tissue kallikrein. Biochemical Journal, 253: 313-321.
- 3. Del Nery E, Chagas JR, Juliano MA, Prado ES & Juliano L (1995). Evaluation of the extent of the binding site in human tissue kallikrein by synthetic substrates with sequences of human kininogen fragments. Biochemical Journal, 312: 233-238.
- 4. Berg T, Bradshaw RA, Carretero OA, Chao J, Chao L, Clements JA, Fahnestock M, Fritz H, Gauthier F, MacDonald RJ, Margolius HS, Morris BJ, Richards RI & Scicli AG (1992). A common nomenclature for members of the tissue (glandular) kallikrein gene families. Agents and Actions, 38 (Suppl): 19-25.
- 5. Schachter M (1980). Kallikreins (kininogenases) - A group of serine proteases with bioregulatory actions. Pharmacological Reviews, 31: 1-17.
- 6. Geiger R & Fritz H (1981). Human urinary kallikrein. Methods in Enzymology, 80: 466-492.
- 7. Antonini E, Ascenzi P, Menegatti E, Bortolotti F & Guarneri M (1982). Catalytic properties of human urinary kallikrein. Biochemistry, 21: 2477-2482.
- 8. Chagas JR, Portaro FCV, Hirata IY, Almeida PC, Juliano MA, Juliano L & Prado ES (1995). Determinants of the unusual cleavage specificity of lysyl-bradykinin-releasing kallikreins. Biochemical Journal, 306: 63-69.
- 9. Hial V, Diniz CR & Mares-Guia M (1974). Purification and properties of a human urinary kallikrein (kininogenase). Biochemistry, 13: 4311-4318.
- 10. Geiger R, Mann K & Bettels T (1977). Isolation of human urinary kallikrein by affinity chromatography. Journal of Clinical Chemistry and Clinical Biochemistry, 15: 479-483.
- 11. Kettner C, Mirabelli C, Pierce JV & Shaw E (1980). Active site mapping of human and rat urinary kallikreins by peptidyl chloromethyl ketones. Archives of Biochemistry and Biophysics, 202: 420-430.
- 12. Miranda TLS, Ramos CHI, Freire RTS, Souza EP, Rogana E, Santoro MM & Figueiredo AFS (1995). Kinetic mechanism of the inhibition of human urinary kallikrein by basic pancreatic trypsin inhibitor. Brazilian Journal of Medical and Biological Research, 28: 505-512.
- 13. Rogana E, Nelson DL & Mares-Guia M (1975). Characterization of the ultraviolet absorption spectra of para-substituted derivatives of benzamidine. Journal of the American Chemical Society, 97: 6844-6848.
- 14. Plowman KM (1972). Inhibitor studies. In: Hume DN, Stork G, King EL, Herschbach DR & People JA (Editors), Enzyme Kinetics McGraw-Hill Book Company, New York, 56-75.
- 15. Cornish-Bowden A (1981). Inhibitors and activators. In: Cornish-Bowden A (Editor), Fundamentals of Enzyme Kinetics Butterworth & Co., Ltd., London, 73-98.
- 16. Cram DJ & Hammond GS (1964). Physical properties. In: Cram DJ & Hammond GS (Editors), Organic Chemistry McGraw-Hill Book Company, New York, 183-225.
- 17. Mares-Guia M & Shaw E (1965). Studies on the active center of trypsin. Journal of Biological Chemistry, 210: 1579-1585.
- 18. Katz BA, Liu B, Barnes M & Springman EB (1998). Crystal structure of recombinant human tissue kallikrein at 2.0 Ĺ resolution. Protein Science, 7: 875-885.
- 19. Ascenzi P, Menegatti E, Guarneri M, Bortolotti F & Antonini E (1982). Catalytic properties of serine proteases. 2. Comparison between human urinary kallikrein and human urokinase, bovine ß-trypsin, bovine thrombin, and bovine a-chymotrypsin. Biochemistry, 21: 2483-2490.
- 20. Schechter I & Berger A (1967). On the size of the active site in proteases. I. Papain. Biochemical and Biophysical Research Communications, 27: 157-162.
- 21. Chen Z & Bode W (1983). Refined 2.5 Ĺ X-ray crystal structure of the complex formed by porcine kallikrein A and the bovine pancreatic trypsin inhibitor. Journal of Molecular Biology, 164: 283-310.
- 22. Weber G (1992). Transfer of proteins to apolar media and the dynamic interactions of proteins and membranes. In: Weber G (Editor), Protein Interactions Routledge, Chapman & Hall, Inc., New York, 161-176.
- 23. Riordan JF, McElvany KD & Borders Jr CL (1976). Arginyl residues: anion recognition sites in enzymes. Science, 195: 884-886.
- 24. Paddlan EA, Davis D, Rudikoft S & Potter EM (1976). Structural basis for the specificity of phosphorylcholine-binding immunoglobulins. Immunochemistry, 13: 945-949.
- 25. Segel IH (1975). Rapid equilibrium partial and mixed-type inhibition. In: Segel IH (Editor), Enzyme Kinetics John Wiley & Sons, New York, 161-226.
- 26. Mares-Guia M, Nelson DL & Rogana E (1977). Electronic effects in the interaction of para-substituted benzamidines with trypsin: the involvement of the P-electronic density at the central atom of the substituent in binding. Journal of the American Chemical Society, 99: 2331-2336.
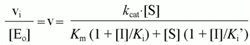
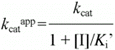
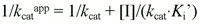
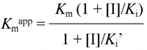
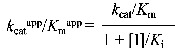
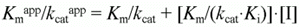
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
08 Jan 2001 -
Date of issue
Jan 2001
History
-
Received
26 Nov 1999 -
Accepted
11 Oct 2000






