
Abstract
The hormone 1,25-dihydroxyvitamin D3 (1,25-(OH)2D3), the active form of vitamin D3, is an important regulator of calcium homeostasis, exerts antiproliferative effects on various cell systems and can induce differentiation in some kinds of hematopoietic cells. These effects are triggered by its receptor, vitamin D receptor (VDR), a phosphoprotein member of the nuclear receptor superfamily, which functions as a transcriptional factor. VDR binds as a heterodimer with retinoid X receptor (R X R) to hexameric repeats, characterized as vitamin D-responsive elements present in the regulatory region of target genes such as osteocalcin, osteopontin, calbindin-D28K, calbindin-D9K, p21WAF1/CIP1, TGF-ß2 and vitamin D 24-hydroxylase. Many factors such as glucocorticoids, estrogens, retinoids, proliferation rate and cell transformation can modulate VDR levels. VDR is expressed in mammary tissue and breast cancer cells, which are potential targets to hormone action. Besides having antiproliferative properties, vitamin D might also reduce the invasiveness of cancer cells and act as an anti-angiogenesis agent. All of these antitumoral features suggest that the properties of vitamin D could be explored for chemopreventive and therapeutic purposes in cancer. However, hypercalcemia is an undesirable side effect associated with pharmacological doses of 1,25-(OH)2D3. Some promising 1,25-(OH)2D3 analogs have been developed, which are less hypercalcemic in spite of being potent antiproliferative agents. They represent a new field of investigation.
Calcitriol; Calcitriol receptor; Calcitriol analog; Breast tumor; Cell proliferation
Braz J Med Biol Res, January 2002, Volume 35(1) 1-9 (Review)
Antiproliferative effects of 1,25-dihydroxyvitamin D 3 on breast cells - A mini review
P. Bortman, M.A.A.K. Folgueira, M.L.H. Katayama, I.M.L. Snitcovsky and M.M. Brentani
Disciplina de Oncologia, Departamento de Radiologia, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil
Text
Abstract
The hormone 1,25-dihydroxyvitamin D3 (1,25-(OH)2D3), the active form of vitamin D3, is an important regulator of calcium homeostasis, exerts antiproliferative effects on various cell systems and can induce differentiation in some kinds of hematopoietic cells. These effects are triggered by its receptor, vitamin D receptor (VDR), a phosphoprotein member of the nuclear receptor superfamily, which functions as a transcriptional factor. VDR binds as a heterodimer with retinoid X receptor (R X R) to hexameric repeats, characterized as vitamin D-responsive elements present in the regulatory region of target genes such as osteocalcin, osteopontin, calbindin-D28K, calbindin-D9K, p21WAF1/CIP1, TGF-ß2 and vitamin D 24-hydroxylase. Many factors such as glucocorticoids, estrogens, retinoids, proliferation rate and cell transformation can modulate VDR levels. VDR is expressed in mammary tissue and breast cancer cells, which are potential targets to hormone action. Besides having antiproliferative properties, vitamin D might also reduce the invasiveness of cancer cells and act as an anti-angiogenesis agent. All of these antitumoral features suggest that the properties of vitamin D could be explored for chemopreventive and therapeutic purposes in cancer. However, hypercalcemia is an undesirable side effect associated with pharmacological doses of 1,25-(OH)2D3. Some promising 1,25-(OH)2D3 analogs have been developed, which are less hypercalcemic in spite of being potent antiproliferative agents. They represent a new field of investigation.
Key words: Calcitriol, Calcitriol receptor, Calcitriol analog, Breast tumor, Cell proliferation

Mechanism of action of vitamin D
Vitamin D is in fact a secosteroid hormone which in its active form, 1,25-dihydroxyvitamin D3 (1,25-(OH)2D3), is an important regulator of bone development and metabolism and calcium homeostasis. Besides these well-known functions on classical target tissues (bone, kidneys, intestine, parathyroids), 1,25-(OH)2D3 plays an important role in the regulation of cell growth and differentiation in cells other than its classical targets.
Vitamin D can be obtained in two distinct ways, i.e., through dietary intake (fatty fish, fish oil, mushrooms, vitamin D-fortified food such as milk) and/or the endogenous pathway, in which the precursor 7-dehydrocholesterol, present in the skin, upon the action of sunlight becomes pre-vitamin D, the latter being subsequently hydroxylated in the liver and kidneys to 25-(OH)D3 and 1,25-(OH)2D3, the active form (1).
The hormone exerts its effects via genomic and non-genomic mechanisms. In the first case, the response is triggered by its nuclear receptor - the vitamin D receptor or VDR, which is a trans-acting transcriptional factor and a member of the nuclear hormone receptor superfamily. The N-terminal domain of VDR is configured into two zinc-coordinated fingers responsible for DNA recognition and binding, whereas the C-terminal domain binds the 1,25-(OH)2D3. VDR binds selectively to DNA primarily as a heterodimer with retinoid X receptor (R X R). The binding of the hormone and the receptor causes a conformational alteration in the linkage domain of the latter with consequent dissociation of co-repressors, facilitating the interactions between VDR and co-activator proteins such as the members of the p160, SRC-1, 2 and 3 families, and the protein complex named DRIP. These co-activators modulate the chromatin structure and the contact with the basal transcriptional factors (2). The receptor/1,25-(OH)2D3 complex regulates gene transcription both positively and negatively through binding motifs in the promoter regions of target genes, designated vitamin D response elements, or VDREs. Several VDREs have been characterized and they generally consist of two direct repeats of six nucleotides (AGGTCA) separated by three aleatory nucleotides; however, neither the sequence of half-sites nor the spacing between them is well conserved. VDREs have been identified in genes such as calbindin-D28K (3) and calbindin-D9K, which are calcium-binding proteins mainly present in mammalian kidney and intestine, respectively; osteocalcin (4) and osteopontin (5), which are bone matrix proteins produced by osteoblasts; vitamin D 24-hydroxylase (6), an enzyme that inactivates 1,25-(OH)2D3; p21WAF1/CIP1 (7), a cyclin-dependent kinase inhibitor; c-fos, an early response gene, and transforming growth factor ß2 (TGF-ß2) (8).
VDR has been detected in numerous classical and nonclassical target tissues of 1,25-(OH)2D3, in tumors of various origins and cell lines such as NIH-3T3 mouse fibroblasts and MCF-7 human breast cancer cells (9,10). The responsiveness of target cells to the hormone depends on the amount of VDR and many factors can modulate VDR levels. Previous studies by our group and others have shown, for example, that glucocorticoids, prolactin, estrogens, retinoids and growth factors might influence VDR expression in mammary and leukemic cells (9,11-14). In HL-60 myeloblastic cells, VDR content correlated indirectly with the proliferation rate expressed by the fraction of cells in the G0/G1 phase (13). However, in other leukemic cell lines such as U937 and K562, phorbol ester treatment caused growth arrest, which was not accompanied by VDR down-regulation (14). Our data for leukemic cells suggest that VDR expression is not consistently changed upon inhibition of cell proliferation.
In addition to this genomic pathway, vitamin D has also been reported to induce nontranscriptional responses involving activation of transmembrane signal transduction pathways. Moreover, 1,25-(OH)2D3 modulates voltage-dependent Ca2+ channel-mediated Ca2+ influx in cultured chick muscle cells by a non-genomic pathway involving G protein-dependent stimulation of both the adenylyl cyclase/cyclic AMP/PKA messenger system and a phosphoinositide-specific phospholipase C (PLC). The rapid activation of PLC, in turn, generates diacylglycerol and inositol-1,4,5-triphosphate, promoting the activation of protein kinase C and rapid release of Ca2+ from endogenous stores (15). These effects could be mediated by a putative but as yet unidentified membrane receptor (16). Other findings support evidence that some actions of 1,25-(OH)2D3 such as monocyte differentiation are mediated by activation of phosphatidylinositol 3-kinase, a lipid kinase, which forms a complex with VDR, suggesting that non-genomic and genomic mechanisms could take place in concert (17).
Vitamin D and breast cancer
Epidemiological studies have shown that death rate and incidence of breast cancer tend to increase with increasing latitude, suggesting that solar radiation might play a protective role in breast cancer development. These studies also provide provocative data indicating an inverse relationship between decreased sunlight exposure and diminished vitamin D production on the skin and higher breast cancer incidence and mortality (18,19). According to this hypothesis, it was reported that white women affected by breast cancer show lower 1,25-(OH)2D3 blood levels than unaffected ones (20). In addition, Mawer et al. (21), in a study on breast cancer patients, found the highest serum 1,25-(OH)2D3 levels in the early stage as compared to more advanced bone metastatic disease.
Another possible link between the vitamin D pathway and breast cancer was recently reported as an amplification of CYP24, located in a region of recurrent aberration at 20q13.2 in breast cancer. This gene encodes vitamin D 24-hydroxylase, an enzyme responsible for 1,25-(OH)2D3 degradation, and its overexpression could lead to abrogation of growth control mediated by vitamin D (22). On the other hand, 25-(OH)D3-1a-hydroxylase, responsible for 25-(OH)D3 activation, was detected in normal human breast as well as in breast carcinoma samples, indicating that both normal and cancerous tissues could be capable of 25-(OH)D3-1a-hydroxylation (23) and local synthesis of 1,25-(OH)2D3.
Receptors for 1,25-(OH)2D3 in MCF-7, a cultured breast cancer cell line, were first shown by Eisman et al. (10) in 1979 and later VDR expression was reported in carcinogen-induced rat mammary tumors as well (24). VDR was also detected in some normal breast tissues such as the mammary gland of pregnant and lactating rabbits but not in virgin rabbits (25).
The antiproliferative effect of 1,25-(OH)2D3 was demonstrated in vitro in MCF-7 cells and other estrogen receptor-positive as well as -negative breast cancer cell lines (11,26). The in vivo antitumor effect of 1,25-(OH)2D3 or its analogs on rat mammary carcinogen (7,12-dimethylbenzanthracene, or N-methyl-N-nitrosourea)-induced tumors was observed as reduced total tumor burden or extended tumor latency and lessened tumor incidence (27,28).
The presence of VDR was also demonstrated in normal human breast tissue (29) and in a large proportion, ranging from 75 to 93%, of breast tumor biopsy specimens, as assessed by specific 1,25-(OH)2D3 binding in tumor extracts (25,30) or positive immunostaining using antibodies to VDR (29,31). In some small series of patients, correlations of breast cancer VDR status with prognosis were conflicting. Freake et al. (30) examined breast cancer samples from 56 patients using a hormone-binding assay, and VDR content (less or more than 8 fmol/mg protein) could not predict a difference in probability of survival. In contrast, Colston et al. (31) found a longer disease-free survival among patients with VDR-positive breast tumors as evaluated by immunocytochemistry.
We determined VDR expression in breast cancer or adjacent normal tissue from 50 Brazilian patients. VDR mRNA was detected in almost all samples examined (96%), i.e., tumoral or non-tumoral adjacent tissues (Figure 1) (32). In immunohistochemical assays, strong VDR staining was observed in the nuclei of breast cancer epithelial cells, and it was less intense in infiltrating fibroblasts (Figure 2). Although 54% of the tumors expressed higher VDR mRNA levels as compared to normal breast tissue, no significant difference was detected. Moreover, we could not establish any correlation between VDR mRNA expression in breast tumor tissue and disease outcome as evaluated by axillary node status (data not shown).
 32P-labeled specific probes. VDR mRNA was detected in normal (N) and tumoral (T) samples as well as in MDA-MB231 (breast cancer) and HB4A (normal breast) human cell lines. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as mRNA load control.
32P-labeled specific probes. VDR mRNA was detected in normal (N) and tumoral (T) samples as well as in MDA-MB231 (breast cancer) and HB4A (normal breast) human cell lines. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as mRNA load control.
[View larger version of this image (71 K GIF file)]

Vitamin D receptor (VDR) expression in breast cancer tissue as determined by immunohistochemistry assay. Strong VDR staining can be observed in the nuclei of epithelial cells. Magnification 600X.
[View larger version of this image (301 K JPG file)]
Our next step was to test the action of vitamin D on normal and transformed mammary cells. HC11 is a spontaneously immortalized lineage derived from the mammary gland of midpregnant BALB/c mice. These cells retain characteristics of normal cells such as growth inhibition by cell contact and are able to differentiate in vitro and synthesize milk proteins (ß-casein) following stimulation with lactogenic hormones. HC11 cells do not express estrogen receptors and lack wild-type p53 (33). On the other hand, HC11 cells transformed with the oncogene Ha-ras (HC11ras) are no longer growth inhibited upon cell contact, do not respond to lactogenic hormones, and are tumorigenic when injected into nude mice (34). Our studies have demonstrated that only HC11 parental cells are growth inhibited upon 1,25-(OH)2D3 treatment, whereas HC11ras cells respond modestly to the hormone. This differential sensitivity seems to reflect the decreased VDR mRNA content of transformed cells as compared to parental cells (35). Our recent data indicate that even though both cell lines present a similar VDR mRNA transcription rate as evaluated by run off assays, VDR mRNA seems to be less stable in HC11ras than in parental cells (36).
We have also determined if less hypercalcemic 1,25-(OH)2D3 analogs, EB1089 (seocalcitol), which presents a double bond in the C-17 side chain, and KH1060, a C-20 epimeric compound (both donated by Dr. Lise Binderup, Leo Pharmaceutical Products, Ballerup, Denmark), exerted antiproliferative effects on HC11 and HC11ras cells. HC11 cells were growth inhibited by both analogs, in contrast to HC11ras cells, as evidenced by the growth curves presented in Figure 3. A lower concentration of KH1060 (1 nM) as compared to 1,25-(OH)2D3 (10 nM) was able to double the duplication time of HC11 cells in a similar way to the parent compound (37).
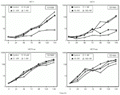 4) were seeded onto 8.8-cm2 plates and maintained in the absence (control) or presence of 1, 10 or 100 nM EB1089 or 10 pm, 0.1 nM or 1 nM KH1060 and harvested at 24-h intervals. Two independent assays were performed in triplicate and mean cell number was plotted on monolog graph paper.
4) were seeded onto 8.8-cm2 plates and maintained in the absence (control) or presence of 1, 10 or 100 nM EB1089 or 10 pm, 0.1 nM or 1 nM KH1060 and harvested at 24-h intervals. Two independent assays were performed in triplicate and mean cell number was plotted on monolog graph paper.
[View larger version of this image (53 K GIF file)]
The inhibitory effects of 1,25-(OH)2D3 on breast cancer cell growth could be mediated by inducing the expression of cyclin-dependent kinase inhibitors such as p21WAF1/CIP1 (38) and p27KIP1 (39). A functional vitamin D response element was described in the promoter region of the p21WAF1/CIP1 gene (7). On the other hand, 1,25-(OH)2D3 positive regulation of the p27KIP1 gene does not directly involve VDR but is mediated by the transcription factors Sp1 and NF-Y (40). Other potential molecular effectors of the antiproliferative actions of 1,25-(OH)2D3 could be TGF-ß1 (41), which exerts antiproliferative actions on epithelial cells and its receptor type II (Tß-RII) (42). In addition, it was shown that the c-myc protooncogene could be down-regulated by 1,25-(OH)2D3 in breast cancer cells (43). The hormone enhances HOXB4 (a homeobox gene product) that binds to MIE1 sites located at intron 1 of the c-myc gene and as a result a transcriptional elongation block takes place (44). It was also demonstrated that 1,25-(OH)2D3 could inhibit the mitogenic activity of insulin and insulin growth factor I-stimulated growth of MCF-7 cells (45) which may be related to insulin growth factor binding protein (IGFBP)-5 (46) and IGFBP-3 (47) up-regulation. Furthermore, the antiproliferative effect of 1,25-(OH)2D3 could be modulated by induction of BRCA1 gene expression, as recently reported (48). On the other hand, 1,25-(OH)2D3-induced growth inhibition may involve activation of apoptosis in vitro with up-regulation of genes associated with mammary gland apoptosis such as TRPM-2/clusterin and cathepsin B as well as with down-regulation of antiapoptotic genes such as bcl-2 (38). Vitamin D-stimulated apoptotic regression in mice bearing MCF-7 xenografts was also described (49).
The underlying mechanism of the antiproliferative effects of 1,25-(OH)2D3 on HC11 cells has yet to be clarified. Our data suggest that it does not involve c-myc down-regulation (Figure 4) (50) or TGF-ß1 up-regulation (51). Cell cycle regulators such as cyclins D1 and D3 and p27KIP1 determined by Western blot were also not involved. We have observed a slight increase of CDKI p21WAF1/CIP1 and cyclin E expression (data not shown).
 myc mRNA in HC11 and HC11ras cells exposed or not (C, control) to 100 nM 1,25-(OH)2D3 (D) for short (2, 4, 6 h) or long (72 h) periods of time, evaluated in Northern blot assays.
myc mRNA in HC11 and HC11ras cells exposed or not (C, control) to 100 nM 1,25-(OH)2D3 (D) for short (2, 4, 6 h) or long (72 h) periods of time, evaluated in Northern blot assays.
[View larger version of this image (67 K GIF file)]
Vitamin D analogs have already been employed in some clinical studies and topical treatment of patients with locally advanced or cutaneous metastatic breast cancer with calcipotriol resulted in very few responses (52), whereas a phase I study in which patients with advanced breast and colorectal cancer received EB1089 showed a few cases of disease stabilization (53).
In addition to having effects on cell proliferation 1,25-(OH)2D3 has been shown to inhibit the invasive potential of breast cancer cells in vitro (54) and its analog EB1089 can prevent skeletal metastasis development in vivo (55) and angiogenesis in vitro and in vivo (56) in human breast carcinoma cells transplanted into nude mice. Furthermore, an interaction between 1,25-(OH)2D3 or analogs with chemotherapeutic drugs active on breast cancer, such as doxorubicin (57) and paclitaxel (58), was also demonstrated, resulting in potentiation of cytotoxicity in MCF-7 cell cultures. In addition, topical 1,25-(OH)2D3 increased the antitumor effect of cyclophosphamide in female mice inoculated with murine mammary tumor (59) and additive effects were observed when a vitamin D analog, CB1093, was administered together with paclitaxel to MCF-7 growing in immunodeficient mice (60).
Taken together, these studies indicate that vitamin D and its analogs have important antitumoral properties, which might be explored in chemopreventive as well as in therapeutic cancer approaches.
Concluding remarks
The involvement of the vitamin D3 pathway in breast carcinogenesis and cancer progression has not been fully clarified. Besides the tumor growth-suppressive activity of vitamin D3 compounds in vitro and in vivo, additional aspects may be involved in the antitumoral properties of this vitamin. Vitamin D3 seems also to induce apoptosis and to inhibit the processes of angiogenesis, invasion and metastasis. Interactions between vitamin D3 and chemotherapeutic drugs have been reported and represent another field to be explored. On the other hand, the pharmacological doses of vitamin D3 necessary to induce antiproliferative effects are associated with hypercalcemia in vivo. New vitamin D3 analogs, which are less hypercalcemic but are potent growth inhibitory agents, might be an option to fight cancer development.
Acknowledgments
The authors want to thank Mrs. Sueli Nonogaki (Instituto Adolfo Lutz, São Paulo, SP, Brazil) who performed the VDR immunohistochemistry assays for Figure 2.
Address for correspondence: M.M. Brentani, Departamento de Radiologia, Faculdade de Medicina, USP, Av. Dr. Arnaldo, 455, 01246-903 São Paulo, SP, Brasil. Fax: +55-11-3082-6580.
Research supported by FAPESP (Nos. 98/16066-9 and 99/00900-2). P. Bortman received the "Young Scientist Luis Renato Caldas-2000" award from the Brazilian Society of Biophysics for this paper. Received February 20, 2001. Accepted October 18, 2001.
- 1. Horst RL & Reinhardt TA (1997). Vitamin D metabolism. In: Feldman D, Glorieux FH & Pike JW (Editors), Vitamin D Academic Press, San Diego, CA, USA.
- 2. Rachez C, Gamble M, Chang CP, Atkins GB, Lazar MA & Freedman LP (2000). The DRIP complex and SRC-1/p160 coactivators share similar nuclear receptor binding determinants but constitute functionally distinct complexes. Molecular and Cellular Biology, 20: 2718-2726.
- 3. Gill RK & Christakos S (1993). Identification of sequence elements in mouse calbindin-D28K gene that confer 1,25-dihydroxyvitamin D3 and butyrate inducible responses. Proceedings of the National Academy of Sciences, USA, 90: 2984-2988.
- 4. Kerner SA, Scott RA & Pike JW (1989). Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3 Proceedings of the National Academy of Sciences, USA, 86: 4455-4459.
- 5. Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF & Denhardt DT (1990). Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (Spp-1 or osteopontin) gene expression. Proceedings of the National Academy of Sciences, USA, 87: 9995-9999.
- 6. Ohyama Y, Noshiro M, Eggertsen G, Gotoh O, Kato Y, Bjorkhem I & Okuda K (1993). Structural characterization of the gene enconding rat 25-hydroxyvitamin D3 24-hydroxylase. Biochemistry, 32: 76-82.
- 7. Liu X, Lee MT, Cohen M, Bormakanti M & Freedman LP (1996). Transcriptional activation of the CDK inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes and Development, 10: 142-153.
- 8. Wu Y, Craig TA, Lutz WH & Kumar R (1999). Identification of 1a,25-dihydroxyvitamin D3 response elements in the human transforming growth factor ß2 gene. Biochemistry, 38: 2654-2660.
- 9. Krishnan AV & Feldman D (1991). Stimulation of 1,25-dihydroxyvitamin D3 receptor gene expression in cultured cells by serum and growth factors. Journal of Bone and Mineral Research, 6: 1099-1107.
- 10. Eisman JA, Martin TJ, MacIntyre I & Moseley JM (1979). 1,25-Dihydroxyvitamin-D-receptor in breast cancer cells. Lancet, 2: 1335-1336.
- 11. Escaleira MTF, Sonohara S & Brentani MM (1993). Sex steroids induced up-regulation of 1,25-(OH)2 vitamin D3 receptors in T47D breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology, 45: 257-263.
- 12. Feldman J, Federico MHH, Sonohara S, Katayama MLH, Koike MAA, Da Silva MRP & Brentani MM (1993). Vitamin D3 binding activity during leukemic cell differentiation. Leukemia Research, 17: 97-101.
- 13. Folgueira MAAK, Federico MHH, Katayama MLH, Silva MRP & Brentani MM (1998). Expression of vitamin D receptor (VDR) in HL-60 cells is differentially regulated during the process of differentiation induced by phorbol ester, retinoic acid or interferon-g. Journal of Steroid Biochemistry and Molecular Biology, 66: 193-201.
- 14. Folgueira MAAK, Federico MHH, Roela RA, Maistro S, Katayama MLH & Brentani MM (2000). Differential regulation of vitamin D receptor expression in distinct leukemic cell lines upon phorbol ester-induced growth arrest. Brazilian Journal of Medical and Biological Research, 33: 559-568.
- 15. Capiati DA, Vazquez G, Iñon MTT & Boland RL (2000). Role of protein kinase C in 1,25(OH)2-vitamin D3 modulation of intracellular calcium during development of skeletal muscle cells in culture. Journal of Cellular Biochemistry, 77: 200-212.
- 16. Nemere I, Dormanen MC, Hammond MW, Okamura WH & Norman AW (1994). Identification of a specific binding protein for 1a,25-dihydroxyvitamin D3 in basal-lateral membranes of chick intestinal epithelium and relationship to transcaltachia. Journal of Biological Chemistry, 269: 23750-23756.
- 17. Hmama Z, Nandan D, Knutson KL, Herrera-Velit P & Reiner NE (1999). 1a,25-Dihydroxyvitamin D3-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. Journal of Experimental Medicine, 190: 1583-1594.
- 18. Garland FC, Garland CF, Gorham ED & Young JF (1990). Geographic variation in breast cancer mortality in the United States: a hypothesis involving exposure to solar radiation. Preventive Medicine, 19: 614-622.
- 19. John EM, Schwartz GG, Dreon DM & Koo J (1999). Vitamin D and breast cancer risk: the NHANES epidemiologic follow-up study, 1971-1975 to 1992. National Health and Nutrition Examination Survey. Cancer Epidemiology, Biomarkers and Prevention, 8: 399-406.
- 20. Janowsky EC, Lester GE, Weinberg CR, Millikan RC, Schildkraut JM, Garrett PA & Hulka BS (1999). Association between low levels of 1,25-dihydroxyvitamin D and breast cancer risk. Public Health Nutrition, 2: 283-291.
- 21. Mawer EB, Walls J, Howell A, Davies M, Ratcliffe WA & Bundred NJ (1997). Serum 1,25-dihydroxyvitamin D may be related inversely to disease activity in breast cancer patients with bone metastases. Journal of Clinical Endocrinology and Metabolism, 82: 118-122.
- 22. Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, Kuo WL, Gray JW & Pinkel D (2000). Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nature Genetics, 25: 144-146.
- 23. Friedrich M, Reichrath J, Chen TC, Tanpricha V, Gherson I, Tilgen W, Schmidt W & Holick MF (2000). Expression of 25-hydroxyvitamin D3-1a-hydroxylase in breast tissue. In: Norman AW, Bouillon R & Thomasset M (Editors), Vitamin D Endocrine System Structural, Biological, Genetic and Clinical Aspects. Proceedings of the Eleventh Workshop on Vitamin D Printing and Reprographics University of California, Riverside, CA, USA, 189-191.
- 24. Colston K, Wilkinson JR & Coombes RC (1986). 1,25-Dihydroxyvitamin D3 binding in estrogen responsive rat breast tumor. Endocrinology, 119: 397-403.
- 25. Eisman JA, MacIntyre I, Martin TJ, Frampton RJ & King RJ (1980). Normal and malignant breast tissue is a target organ for 1,25-(OH)2 vitamin D3 Clinical Endocrinology, 13: 267-272.
- 26. Chouvet C, Vicard E, Devonec M & Saez S (1986). 1,25-Dihydroxyvitamin D3 inhibitory effect on the growth of two human breast cancer cell lines (MCF-7, BT-20). Journal of Steroid Biochemistry and Molecular Biology, 24: 373-376.
- 27. Iino Y, Hyoshida M, Sugamata N, Maemura M, Ohwada S, Yokoe T, Ishikita T, Horiuchi R & Morishita Y (1992). 1Alpha-hydroxyvitamin D3, hypercalcemia, and growth suppression of 7,12-dimethylbenz(a)anthracene-induced rat mammary tumors. Breast Cancer Research and Treatment, 22: 133-140.
- 28. Mehta R, Hawthorne M, Uselding L, Albinescu D, Moriarty R, Christov K & Mehta R (2000). Prevention of N-methyl-N-nitrosourea-induced mammary carcinogenesis in rats by 1 alpha-hydroxyvitamin D(5). Journal of the National Cancer Institute, 92: 1836-1840.
- 29. Friedrich M, Rafi L, Tilgen W, Schmidt W & Reichrath JV (1998). Expression of 1,25-dihydroxyvitamin D3 receptor in breast carcinoma. Journal of Histochemistry and Cytochemistry, 46: 1335-1337.
- 30. Freake HC, Abeyasekera G, Iwasaki J, Marocci C, MacIntyre I, McClelland RA, Skilton RA, Easton DF & Coombes RC (1984). Measurement of 1,25-dihydroxyvitamin D3 receptors in breast cancer and relationship to biochemical and clinical indices. Cancer Research, 44: 1677-1681.
- 31. Colston KW, Berger U & Coombes RC (1989). Possible role for vitamin D in controlling breast cancer cell proliferation. Lancet, 1: 185-191.
- 32. Katayama MLH, Barboza EM & Brentani MM (2000). Expression of vitamin D receptor and 24-hydroxylase genes in breast cancer. Cancer Detection and Prevention, 24: S126.
- 33. Merlo GR, Venesio T, Taverna D, Marte BM, Callahan R & Hynes NE (1994). Growth suppression of normal mammary epithelial cells by wild type p53. Oncogene, 9: 443-453.
- 34. Hynes NE, Taverna D, Harweth IM, Clardiello F, Salomon DS, Yamamoto T & Groner B (1990). Epidermal growth factor receptor, but not c-erb-2, activation prevents lactogenic hormone induction of ß-casein gene in mouse mammary epithelial cells. Molecular and Cellular Biology, 10: 4027-4043.
- 35. Escaleira MTF & Brentani MM (1999). Normal x Ha-ras-transformed HC11 mouse mammary cells: different responses to the antiproliferative activity of vitamin D3 and modulation of its receptor. Breast Cancer Research and Treatment, 54: 123-134.
- 36. Bortman P, Folgueira MAAK, Katayama MLH, Snitcovsky IML & Brentani MM (2000). Regulação da expressão de RNAm de VDR e caracterização do envolvimento da via genômica e não genômica da vitamina D em células HC11 parentais ou transformadas com H-ras. Anais of the XV Annual Meeting of the Federação de Sociedades de Biologia Experimental, Caxambu, MG, Brazil, August 23-26, 2000, 98-99.
- 37. Katayama MLH, Folgueira MAAK, Snitcovsky IML, Garcia EF, Apolinário D, Bortman P, Roela RA & Brentani MM (2000). HC11 mouse mammary cells are growth inhibited by EB1089 and KH1060 with a late induction of TGF-ß expression while HC11 Ha-ras transformed cells are not affected by these compounds. In: Norman AW, Bouillon R & Thomasset M (Editors), Vitamin D Endocrine System Structural, Biological, Genetic and Clinical Aspects. Proceedings of the Eleventh Workshop on Vitamin D. Printing and Reprographics University of California, Riverside, CA, USA, 407-410.
- 38. James SY, Mackay AG & Colston KW (1996). Effects of 1,25 dihydroxyvitamin D3 and its analogues on induction of apoptosis in breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology, 58: 395-401.
- 39. Wu G, Fan RS, Li W, Ko TC & Brattain MG (1997). Modulation of cell cycle control by vitamin D3 and its analogue, EB 1089, in human breast cancer cells. Oncogene, 15: 1555-1563.
- 40. Inoue T, Kamiyama J & Sakai T (1999). Sp1 and NF-Y synergistically mediate the effect of vitamin D3 in the p27Kip1 gene promoter that lacks vitamin D response elements. Journal of Biological Chemistry, 274: 32309-32317.
- 41. Koli K & Keski-Oja J (1995). 1,25-Dihydroxyvitamin D3 enhances the expression of transforming growth factor beta 1 and its latent form binding protein in cultured breast carcinoma cells. Cancer Research, 55: 1540-1546.
- 42. Wu G, Fan RS, Li W, Srinivas V & Brattain MG (1998). Regulation of transforming growth factor-beta type II receptor expression in human breast cancer MCF-7 cells by vitamin D3 and its analogs. Journal of Biological Chemistry, 273: 7749-7756.
- 43. Saunders DE, Christensen C, Wappler NL, Schultz JF, Lawrence WD, Malviya VK, Malone JM & Deppe G (1993). Inhibition of c-myc in breast and ovarian carcinoma cells by 1,25-dihydroxyvitamin D3, retinoic acid and dexamethasone. Anticancer Drugs, 4: 201-208.
- 44. Pan Q & Simpson RU (2001). Antisense knockout of HOXB4 blocks 1,25-dihydroxyvitamin D3 inhibition of c-myc expression. Journal of Endocrinology, 169: 153-159.
- 45. Vink-van Wijngaarden T, Pols HA, Buurman CJ, Birkenhager JC & van Leeuwen JP (1996). Inhibition of insulin- and insulin-like growth factor-I-stimulated growth of human breast cancer cells by 1,25-dihydroxyvitamin D3 and the vitamin D3 analogue EB1089. European Journal of Cancer, 32A: 842-848.
- 46. Rozen F, Yang XF, Huynh H & Pollak M (1997). Antiproliferative action of vitamin D-related compounds and insulin-like growth-factor-binding protein 5 accumulation. Journal of the National Cancer Institute, 89: 652-656.
- 47. Colston KW, Perks CM, Xie SP & Holly JM (1998). Growth inhibition of both MCF-7 and Hs578T human breast cancer cell lines by vitamin D analogues is associated with increased expression of insulin-like growth factor binding protein-3. Journal of Molecular Endocrinology, 20: 157-162.
- 48. Campbell MJ, Gombart AF, Kwok SH, Park S & Koeffler HP (2000). The anti-proliferative effects of 1alpha,25(OH)2D3 on breast and prostate cancer cells are associated with induction of BRCA gene expression. Oncogene, 19: 5091-5097.
- 49. VanWeelden K, Flanagan L, Binderup L, Tenniswood M & Welsh JW (1998). Apoptotic regression of MCF-7 xenografts in nude mice treated with the vitamin D3 analog, EB1089. Endocrinology, 139: 2102-2110.
- 50. Folgueira MAAK, Katayama MLH, Snitcovsky IML & Brentani MM (1998). c-myc and c-max mRNA expression in HC11 mouse mammary cells exposed to 1,25(OH)2D3 are events unrelated to growth arrest. In: Moraes M, Brentani R & Bevilacqua R (Editors), 17th International Cancer Congress Monduzzi Editore, Bologna, Italy.
- 51. Folgueira MAAK, Katayama MLH, Snitcovsky IML & Brentani MM (2000). The antiproliferative effect of 1,25(OH)2D3 on HC11 mammary cells is not associated to induction of TGFß and p21WAF1/CIP1 or inhibition of c-myc expression. Breast Cancer Research and Treatment, 64: 115 (Abstract).
- 52. Bower M, Colston KW, Stein RC, Hedley A, Gazet JC, Ford HT & Coombes RC (1991). Topical calcipotriol treatment in advanced breast cancer. Lancet, 337: 701-702.
- 53. Gulliford T, English J, Colston KW, Mendy P, Moller S & Coombes RC (1998). A phase I study of the vitamin D analogue EB 1089 in patients with advanced breast and colorectal cancer. British Journal of Cancer, 78: 6-13.
- 54. Hansen CM, Frandsen TL, Brünner N & Binderup L (1994). 1,25-Dihydroxyvitamin D3 inhibits the invasive potential of human breast cancer cells in vitro Clinical and Experimental Metastasis, 12: 195-202.
- 55. El Abdaimi K, Dion N, Papavasiliou V, Cardinal PE, Binderup L, Goltzman D, Ste-Marie LG & Kremer R (2000). The vitamin D analogue EB 1089 prevents skeletal metastasis and prolongs survival time in nude mice transplanted with human breast cancer cells. Cancer Research, 60: 4412-4418.
- 56. Mantell DJ, Owens PE, Bundred NJ, Mawer EB & Canfield AE (2000). 1Alpha,25-dihydroxyvitamin D3 inhibits angiogenesis in vitro and in vivo Circulation Research, 87: 214-220.
- 57. Chaudhry M, Sundaram S, Gennings C, Carter H & Gewirtz DA (2001). The vitamin D3 analog, ILX-23-7553, enhances the response to adriamycin and irradiation in MCF-7 breast tumor cells. Cancer Chemotherapy and Pharmacology, 47: 429-436.
- 58. Wang Q, Yang W, Uytingco MS, Christakos S & Wieder R (2000). 1,25-Dihydroxyvitamin D3 and all-trans-retinoic acid sensitize breast cancer cells to chemotherapy-induced cell death. Cancer Research, 60: 2040-2048.
- 59. Chen G, Baechle A, Nevins TD, Oh S, Harmon C & Stacey DW (1998). Protection against cyclophosphamide-induced alopecia and inhibition of tumor growth by topical 1,25-dihydroxyvitamin D3 in mice. International Journal of Cancer, 2: 303-309.
- 60. Koshizuka K, Koike M, Kubota T, Said J, Binderup L & Koeffler HP (1998). Novel vitamin D3 analog (CB1093) when combined with paclitaxel and cisplatin inhibit growth of MCF-7 human breast cancer cells in vivo International Journal of Oncology, 3: 421-428.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
10 Dec 2001 -
Date of issue
Jan 2002
History
-
Received
20 Feb 2001 -
Accepted
18 Oct 2001
