
Abstract
We evaluated the effects of angiotensin-(1-7) (Ang-(1-7)) on post-ischemic function in isolated hearts from adult male Wistar rats perfused according to the Langendorff technique. Local ischemia was induced by coronary ligation for 15 min. After ischemia, hearts were reperfused for 30 min. Addition of angiotensin II (Ang II) (0.20 nM, N = 10) or Ang-(1-7) (0.22 nM, N = 10) to the Krebs-Ringer perfusion solution (KRS) before the occlusion did not modify diastolic or systolic tension, heart rate or coronary flow (basal values for Ang-(1-7)-treated hearts: 0.72 ± 0.08 g, 10.50 ± 0.66 g, 216 ± 9 bpm, 5.78 ± 0.60 ml/min, respectively). During the period of occlusion, the coronary flow, heart rate and systolic tension decreased (values for Ang-(1-7)-treated hearts: 2.83 ± 0.24 ml/min, 186 ± 7 bpm, 6.95 ± 0.45 g, respectively). During reperfusion a further decrease in systolic tension was observed in control (4.95 ± 0.60 g) and Ang II-treated hearts (4.35 ± 0.62 g). However, in isolated hearts perfused with KRS containing Ang-(1-7) the further reduction of systolic tension during the reperfusion period was prevented (7.37 ± 0.68 g). The effect of Ang-(1-7) on the systolic tension was blocked by the selective Ang-(1-7) antagonist A-779 (2 nM, N = 9), by the bradykinin B2 antagonist HOE 140 (100 nM, N = 10), and by indomethacin pretreatment (5 mg/kg, ip, N = 8). Pretreatment with L-NAME (30 mg/kg, ip, N = 8) did not change the effect of Ang-(1-7) on systolic tension (6.85 ± 0.61 g). These results show that Ang-(1-7) at low concentration (0.22 nM) improves myocardial function (systolic tension) in ischemia/reperfusion through a receptor-mediated mechanism involving release of bradykinin and prostaglandins.
Angiotensin-(1-7); Rat heart; Ischemia/reperfusion; Systolic and diastolic tension
Braz J Med Biol Res, September 2002, Volume 35(9) 1083-1090
Angiotensin-(1-7) improves the post-ischemic function in isolated perfused rat hearts
A.J. Ferreira, R.A.S. Santos and A.P. Almeida
Laboratório de Hipertensão, Departamento de Fisiologia e Biofísica, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brasil
References
Abstract
We evaluated the effects of angiotensin-(1-7) (Ang-(1-7)) on post-ischemic function in isolated hearts from adult male Wistar rats perfused according to the Langendorff technique. Local ischemia was induced by coronary ligation for 15 min. After ischemia, hearts were reperfused for 30 min. Addition of angiotensin II (Ang II) (0.20 nM, N = 10) or Ang-(1-7) (0.22 nM, N = 10) to the Krebs-Ringer perfusion solution (KRS) before the occlusion did not modify diastolic or systolic tension, heart rate or coronary flow (basal values for Ang-(1-7)-treated hearts: 0.72 ± 0.08 g, 10.50 ± 0.66 g, 216 ± 9 bpm, 5.78 ± 0.60 ml/min, respectively). During the period of occlusion, the coronary flow, heart rate and systolic tension decreased (values for Ang-(1-7)-treated hearts: 2.83 ± 0.24 ml/min, 186 ± 7 bpm, 6.95 ± 0.45 g, respectively). During reperfusion a further decrease in systolic tension was observed in control (4.95 ± 0.60 g) and Ang II-treated hearts (4.35 ± 0.62 g). However, in isolated hearts perfused with KRS containing Ang-(1-7) the further reduction of systolic tension during the reperfusion period was prevented (7.37 ± 0.68 g). The effect of Ang-(1-7) on the systolic tension was blocked by the selective Ang-(1-7) antagonist A-779 (2 nM, N = 9), by the bradykinin B2 antagonist HOE 140 (100 nM, N = 10), and by indomethacin pretreatment (5 mg/kg, ip, N = 8). Pretreatment with L-NAME (30 mg/kg, ip, N = 8) did not change the effect of Ang-(1-7) on systolic tension (6.85 ± 0.61 g). These results show that Ang-(1-7) at low concentration (0.22 nM) improves myocardial function (systolic tension) in ischemia/reperfusion through a receptor-mediated mechanism involving release of bradykinin and prostaglandins.
Key words: Angiotensin-(1-7), Rat heart, Ischemia/reperfusion, Systolic and diastolic tension
Introduction
Components of the renin-angiotensin system (RAS) have been identified by molecular biology and biochemical techniques in many tissues, leading to the concept of tissue RAS (1-3) or more properly, local angiotensin-forming systems. Thus, the RAS is viewed now not only as an endocrine system but also as an autocrine/paracrine modulator of tissue functions (heart, blood vessels, kidney, brain and endocrine glands) (1,3-7). The major component of the RAS is the octapeptide angiotensin II (Ang II). In the heart, Ang II induces direct positive inotropic and chronotropic effects on the cardiac muscle, as well as the alteration of cardiac metabolism and vasoconstriction of coronary blood vessels (8). Ang II receptors have been detected throughout the rat heart, with high density in cardiac nerves and lower levels associated with the atria, ventricles and vasculature (9). Ang II receptors are up-regulated following myocardial infarction and hypertrophy, but down-regulated in end-stage heart failure (8,10,11).
In addition to Ang II, other Ang I fragments are active (6,12-15). Angiotensin-(1-7) (Ang-(1-7)) is now considered to be a RAS hormone (6,14,15). It appears to counterbalance actions of Ang II, acting on the cardiovascular system, kidneys and central nervous system (6,12-14). Ang-(1-7) is involved in blood pressure regulation and presents antiproliferative and antithrombogenic effects (6,12,13). We have shown that Ang I is metabolized in the rat coronary circulation (Langendorff preparation) to produce several biologically active angiotensins: Ang II, Ang III, Ang-(3-8) and Ang-(1-7) (16). Formation of Ang II and its carboxyl terminal fragments is partially dependent upon angiotensin-converting enzyme, while the formation of Ang-(1-7) is not modified significantly by angiotensin-converting enzyme inhibitors (16). We have also demonstrated in hearts perfused with Krebs-Ringer solution (KRS) that Ang-(1-7) produced a concentration-dependent (27-210 nM) reduction in coronary flow (25% reduction at highest concentration), while only slight and variable changes in contraction force and heart rate were observed. Under the same conditions, Ang II (27 and 70 nM) produced a significant reduction in coronary flow (39 and 48%, respectively) associated with a significant increase in force (17). In contrast, Almeida et al. (18) have shown that in isolated rat hearts Ang-(1-7) induced an increase in the vasodilator effect of bradykinin (BK) through a nitric oxide (NO) and prostaglandin release-related mechanism.
Recently, we have shown that at a low concentration Ang-(1-7) decreased the incidence and duration of ischemia/reperfusion arrhythmias in isolated rat hearts. These cardioprotective effects were blocked by the Ang-(1-7) antagonist A-779 (19) and by indomethacin pretreatment, but not by the BK-B2 antagonist HOE 140 or by L-NAME pretreatment (20). In the present study we extended this observation by examining the effects of Ang-(1-7) on the post-ischemic function of isolated rat hearts.
Material and Methods
Male Wistar rats (200-300 g body weight) were decapitated 10-15 min after intraperitoneal injection of 400 IU heparin. The thorax was opened and the heart was carefully dissected and perfused through a 1.0 ± 0.3 cm aortic stump with KRS containing 118.4 mM NaCl, 4.7 mM KCl, 1.2 KH2PO4, 1.2 mM MgSO4.7 H2O, 2.5 mM CaCl2.2 H2O, 11.7 mM glucose, and 26.5 mM NaHCO3. The perfusion fluid was maintained at 37 ± 1ºC, with a pressure of 65 mmHg and constant oxygenation (5% CO2 and 95% O2). A force transducer (model FT 03, Grass, West Warwick, RI, USA) was attached through a heart clip to the apex of the ventricles to record the contractile force (tension, g) on a computer using a data acquisition system (Codas, Dataq Instruments, Inc., Akron, OH, USA). A diastolic tension of 0.5 to 1.0 g was applied to the hearts. Electrical activity was recorded with an electrocardiograph (Nihon Kohden, Tokyo, Japan) with the aid of two cotton wicks placed directly on the surface of the right atrium and left ventricle (bipolar lead). Heart rate was calculated from the electrocardiographic records and coronary flow was measured by collecting the perfusate over a period of 1 min at regular intervals. The hearts were perfused for an initial 30-min period with 1) KRS [control, N = 10], or KRS containing 2) Ang II [0.20 nM, N = 10], 3) Ang-(1-7) [0.22 nM, N = 10], 4) A-779 [2 nM, N = 9], 5) A-779 [2 nM] plus Ang-(1-7) [0.22 nM, N = 9], 6) HOE 140 [100 nM, N = 6], and 7) HOE 140 [100 nM] plus Ang-(1-7) [0.22 nM, N = 10]. After the equilibration period the left anterior descending coronary artery was ligated by the method described by Lubbe et al. (21) beneath the left auricular appendage together with the adjacent veins. The ligature was released after 15 min and reperfusion with different KRS (above) was performed for an additional 30 min.
In order to evaluate the role of cyclo-oxygenase products in the effects of Ang-(1-7), rats received indomethacin (5 mg/kg, ip) plus heparin (400 IU, ip). After 1 h the rats were decapitated, the thorax was opened and the heart was dissected and perfused with KRS or KRS containing Ang-(1-7) (0.22 nM, N = 8). After the equilibration period, the left anterior descending coronary artery was ligated as described above. In another experimental group, we determined the role of NO in the effects of Ang-(1-7). Rats received L-NAME (30 mg/kg, ip) plus heparin (400 IU, ip, N = 8). The protocol was the same as that described above. All experimental protocols were performed in accordance with the guidelines for the humane use of laboratory animals of our institute and approved by local authorities.
Statistical analysis
Data are reported as mean ± SEM. Statistical analysis was performed by the Student t-test or by two-way ANOVA followed by the Bonferroni test. P<0.05 was considered to be significant.
Results
Figure 1 shows the changes in systolic and diastolic tension before, during and after occlusion. During the period of occlusion the systolic tension decreased significantly in all groups (Figure 1A). Similar alterations were observed for the dT/dt (Table 1). During reperfusion there was a further decrease in systolic tension and dT/dt in the control and Ang II-treated hearts. However, in the hearts perfused with KRS containing Ang-(1-7) the further decrease in systolic tension or dT/dt was not observed (Figure 1A and Table 1, respectively). As observed for systolic function, only in the hearts perfused with Ang-(1-7) was the velocity of relaxation (-dT/dt) preserved after reperfusion (Table 1).
As shown in Figure 2A, the effect of Ang-(1-7) on systolic tension was significantly blocked by A-779 (2 nM, N = 9), by the BK-B2 antagonist HOE 140 (100 nM, N = 10), and by indomethacin pretreatment (5 mg/kg, ip, N = 8). The increase in diastolic tension after the period of reperfusion was not changed by A-779 or HOE 140 treatment. A further increase in diastolic tension was observed in hearts pretreated with indomethacin and perfused with KRS containing Ang-(1-7) (Figure 2B).
The participation of prostaglandin and NO in the effects induced by Ang-(1-7) was investigated by pretreatment with indomethacin or a NO synthase inhibitor. Figure 3 shows the changes in coronary flow in isolated hearts perfused with normal KRS or KRS containing Ang-(1-7) and the effect of pretreatment with indomethacin or L-NAME on the response of coronary flow to Ang-(1-7). Coronary flow did not change before the period of occlusion. Occlusion of the coronary artery resulted in a comparable flow reduction (approximately 50%), which was sustained throughout the ischemic period. In hearts taken from rats pretreated with L-NAME and perfused with KRS containing Ang-(1-7) a significantly decrease in coronary flow was observed during the reperfusion period. In contrast, in hearts from rats pretreated with indomethacin and perfused or not with Ang-(1-7) an increase in coronary flow was observed. The other treatments alone or in combination with Ang-(1-7) did not change coronary flow.
The heart rate of the control group (basal value, 216 ± 9 bpm) did not differ during basal, occlusion and reperfusion conditions from that of Ang-(1-7)-treated hearts (data not shown).
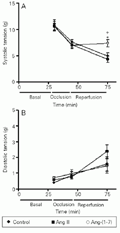
Time course of systolic (A) and diastolic tension (B) in isolated rat hearts. The hearts were perfused with Krebs-Ringer solution (KRS, control, N = 10), KRS containing 0.22 nM Ang-(1-7) (N = 10) or KRS containing 0.20 nM Ang II (N = 10) before and after (reperfusion) coronary occlusion. The maneuvers are indicated on the abscissa. *P<0.05 compared to the control group and
[View larger version of this image (24 K GIF file)]
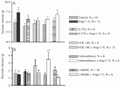 ip, N = 8) or L-NAME (30 mg/kg, ip, N = 8) was administered 1 h before the animals were killed and hearts collected. The hearts were perfused with normal Krebs-Ringer solution (KRS) or KRS containing 0.22 nM Ang-(1-7). *P<0.05 vs untreated hearts and +P<0.05 vs respective control group (Student t-test).
ip, N = 8) or L-NAME (30 mg/kg, ip, N = 8) was administered 1 h before the animals were killed and hearts collected. The hearts were perfused with normal Krebs-Ringer solution (KRS) or KRS containing 0.22 nM Ang-(1-7). *P<0.05 vs untreated hearts and +P<0.05 vs respective control group (Student t-test).
[View larger version of this image (48 K GIF file)]
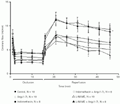 ip, N = 8) and L-NAME (30 mg/kg, ip, N = 8) were administered 1 h before animal sacrifice and heart collection. The hearts were perfused with normal Krebs-Ringer solution (KRS) or KRS containing 0.22 nM Ang-(1-7). *P<0.05 vs control group (two-way ANOVA followed by Bonferroni test).
ip, N = 8) and L-NAME (30 mg/kg, ip, N = 8) were administered 1 h before animal sacrifice and heart collection. The hearts were perfused with normal Krebs-Ringer solution (KRS) or KRS containing 0.22 nM Ang-(1-7). *P<0.05 vs control group (two-way ANOVA followed by Bonferroni test).
[View larger version of this image (36 K GIF file)]
Discussion
The present study was carried out to examine the effect of Ang-(1-7) on the deleterious effects of ischemia and reperfusion on cardiac contractile function and heart rate in a model of 15-min occlusion/ischemia followed by 30-min reperfusion. Ang-(1-7) (0.22 nM) substantially improved myocardial function in ischemia/reperfusion, mainly by preventing the decrease in systolic tension usually observed after reperfusion, without changing heart rate. In addition, during reperfusion no further decrease was observed in +dT/dt or -dT/dt in isolated rat hearts perfused with Ang-(1-7), indicating a beneficial effect of Ang-(1-7) on post-ischemic contractile function. The beneficial effect of Ang-(1-7) on systolic tension was blocked by the Ang-(1-7) antagonist A-779, the BK-B2 antagonist HOE 140 and indomethacin, but not by L-NAME.
The effects of Ang-(1-7) on systolic tension and dT/dt were completely blocked by the Ang-(1-7) antagonist A-779. This finding is consistent with our observations on isolated hearts (18,20) and other preparations (for a review, see Ref. 13). These data suggest that, as observed in other organs, the effects of Ang-(1-7) on the rat heart are receptor-mediated, involving a single Ang-(1-7)-binding site which can be blocked by its analogue A-779 (D-Ala7-Ang-(1-7)) (13,19).
In isolated hearts perfused with KRS containing A-779 we observed a small decrease in systolic tension. However, this change was not statistically significant. The absence of a significant effect of A-779 is not unexpected, even assuming a role for endogenous Ang-(1-7) in this situation, if we consider the multifactorial characteristics of the mechanisms involved in the mechanical changes induced by ischemia/reperfusion (22-24).
There are several mechanisms that might be responsible for the beneficial effects of Ang-(1-7) on systolic function during ischemia and subsequent reperfusion. We and others have shown that 1) Ang-(1-7) at relatively low concentration (2.2 nM) increased the BK-induced vasodilator responses through release of NO and vasodilator prostaglandins (18), 2) Ang-(1-7) present in the perfusion solution at 0.2 nM concentration reduced the incidence and duration of reperfusion arrhythmias (20), 3) Ang-(1-7) exerted some of its effects by releasing BK through a still unknown mechanism (13), and 4) Ang-(1-7) promoted the release of prostanoids and NO (13). In a previous study concerning the protective effects of Ang-(1-7) on reperfusion arrhythmias we observed that HOE 140 had no effect on the antiarrhythmogenic effect of Ang-(1-7). This is in sharp contrast with the complete blockade of the effects of Ang-(1-7) on contractile function by HOE 140. However, as observed in our previous study, pretreatment with L-NAME did not block the effect of Ang-(1-7). These observations suggest that the beneficial effect of Ang-(1-7) on post-ischemic heart contractile function, but not on reperfusion arrhythmias, is mediated by a BK-dependent, NO-independent mechanism. Indeed, Pabla and Curtis (25) showed that endogenous NO does not appear to facilitate early recovery from systolic and diastolic stunning as a result of any direct action on the myocardium. The role of NO in ischemia/reperfusion appears to be dependent on the time of ischemia (26), being more significant with longer periods of coronary occlusion (>35 min). One may argue that L-NAME had no effect because it was washed out from the isolated heart. However, we have previously shown that, under the same conditions as used in the present study, L-NAME completely blocked the BK-potentiating activity of Ang-(1-7) (18). Taken together, our results suggest that the effects of Ang-(1-7) on systolic function were due to a receptor-mediated release and/or potentiation of BK with subsequent release of prostanoids (13). This hypothesis is supported by the effective blockade of the effect of Ang-(1-7) by A-779 and indomethacin.
Isolated hearts obtained from indomethacin-treated rats did not show any significant change in pre- or post-ischemic myocardial mechanical function. However, in indomethacin-treated hearts Ang-(1-7) induced a marked increase in diastolic tension, illustrating the cardioprotective effect of prostaglandins on post-ischemic myocardial function probably mediated by E-type prostanoid3 receptors (27). Formation of toxic oxygen radicals, Ca2+ loading, uncoupling of mitochondrial respiration and mechanical injury to the sarcolemma (22-24) are mechanisms proposed to account for reperfusion-induced cell dysfunction. Further studies are required to clarify which of these mechanisms are involved in this deleterious effect of Ang-(1-7) in the presence of prostaglandin synthesis blockade.
Myocardial ischemia is associated with an altered formation and release of arachidonic acid and its metabolites (27). Thromboxane A2 is a potent coronary vasoconstrictor (28) and contributes to the genesis of arrhythmias (29) and cyclical reductions in coronary blood flow (30) in the ischemic myocardium. Hydroxyeicosatetraenoic acid and thromboxane A2 appear to contribute to the "no-reflow" phenomenon after reperfusion of previously ischemic myocardium (31). On the other hand, Zi et al. (28) found that thromboxane A2 has a specific coronary vasospastic action without a direct inotropic effect. Furthermore, the effects of the hydroperoxides are blocked by indomethacin (32). These observations are consistent with our observation that indomethacin pretreatment increased the coronary flow in isolated rat hearts perfused with normal KRS or KRS containing Ang-(1-7).
Whether release of vasoconstrictor prostaglandins and predominance of its effects are involved in the reduction of coronary flow by Ang-(1-7) in isolated hearts taken from L-NAME-treated rats remains to be determined. Release of vasoconstrictor prostaglandins by Ang-(1-7) in the rat coronary circulation has been suggested, although higher concentrations of this peptide were used (17).
Ang II at a concentration similar to that used for Ang-(1-7) (0.20 nM) did not change the basal or post-ischemic inotropism or heart rate. Contrasting results have been published about the inotropic effect of Ang II, including data on the rat (33-39). Ang II induces a positive inotropic response in most species, which is not fully mediated by the ß-adrenergic system (33-35). In the rat Ang II had either no effect or a negative inotropic effect (36,37,39). Traquandi and Riva (37) found that Ang II induced a dose-dependent negative inotropic response in isolated rat hearts. Yoshiyama et al. (39) reported that exogenous Ang II applied to isolated rat hearts before ischemia had deleterious effects on coronary flow, post-ischemic cardiac function and release of creatine kinase. However, Ford et al. (38) recently reported that Ang II reduces infarct size in a concentration-dependent manner and has no effect on contractile stunning associated with ischemia/reperfusion in isolated rat hearts. Further studies are needed to understand the effects of Ang II on the ischemia/reperfusion model.
In addition to reducing the incidence and duration of cardiac arrhythmias, Ang-(1-7) ameliorates the post-ischemic contractile function by a mechanism apparently involving a receptor-mediated release of BK and prostaglandins. This effect may be relevant to explain why cardiac function improves in heart failure after blockade of the RAS in hypertensive patients (40).
Acknowledgments
The authors wish to thank Elizabete Dias Bontempo for skillful technical assistance.
Address for correspondence: A.P. Almeida, Departamento de Fisiologia e Biofísica, ICB, UFMG, Av. Antônio Carlos, 6627, 31270-901 Belo Horizonte, MG, Brasil. Fax: +55-31-3499-2924. E-mail: apa@mono.icb.ufmg.br
Presented at the IV International Symposium on Vasoactive Peptides, Belo Horizonte, MG, Brazil, October 19-21, 2001. Research supported by PRONEX, CNPq and FAPEMIG. Received January 3, 2002. Accepted August 12, 2002.
- 1. Dzau VJ (1988). Molecular and physiological aspects of tissue renin-angiotensin system: emphasis on cardiovascular control. Journal of Hypertension, 6 (Suppl 3): S7-S12.
- 2. Phillips MI, Speakman EA & Kimura B (1992). Levels of angiotensin and molecular biology of the tissue renin-angiotensin systems. Regulatory Peptides, 43: 1-20.
- 3. Wollert KC & Drexler H (1999). The renin-angiotensin system and experimental heart failure. Cardiovascular Research, 43: 838-849.
- 4. Griendling KK, Lassègue B & Alexander W (1996). Angiotensin receptors and their therapeutic implications. Annual Review of Pharmacology and Toxicology, 36: 281-306.
- 5. Baltatu O, Bader M & Ganten D (1998). Functional testing of components of the brain renin-angiotensin system in transgenic animals. In: Ulfendahl H & Aurell M (Editors), Renin-Angiotensin Portland Press, London, England, 105-114.
- 6. Ferrario CM, Chappell MC, Dean RH & Iyer SN (1998). Novel angiotensin peptides regulate blood pressure, endothelial function, and natriuresis. Journal of the American Society of Nephrology, 9: 1716-1722.
- 7. Kim S & Iwao H (2000). Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacological Reviews, 52: 11-34.
- 8. Dostal DE, Hunt RA, Kule CE, Bhat GJ, Karoor V, McWhinney CD & Baker KM (1997). Molecular mechanisms of angiotensin II in modulating cardiac function: intracardiac effects and signal transduction pathway. Journal of Molecular and Cellular Cardiology, 29: 2893-2902.
- 9. Zhou J, Allen AM, Yamada H, Sun Y & Mendelsohn FAO (1994). Localization and properties of the angiotensin-converting enzyme and angiotensin receptors in the heart. In: Lindpainter K & Ganten D (Editors), The Cardiac Renin Angiotensin System. Futura, New York, NY, USA, 63-88.
- 10. Suzuki J, Matsubara H, Urakami M & Inada M (1993). Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Circulation Research, 73: 439-447.
- 11. Meggs LG, Coupet J, Huang H, Li WCP, Capasso JM, Homcy CJ & Anversa P (1993). Regulation of angiotensin II receptors on ventricular myocytes after myocardial infarction in rats. Circulation Research, 72: 1149-1162.
- 12. Buczko W & Kucharewicz I (2000). Angiotensin-(1-7). One step forward? Polish Journal of Pharmacology, 52: 75-81.
- 13. Santos RAS, Campagnole-Santos MJ & Andrade SP (2000). Angiotensin-(1-7): an update. Regulatory Peptides, 91: 45-62.
- 14. Bernstein KE (2002). Two ACEs and a heart. Nature, 417: 799-802.
- 15. Schmaier AH (2002). The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. Journal of Clinical Investigation, 109: 1007-1009.
- 16. Neves LAA, Almeida AP, Khosla MC & Santos RAS (1995). Metabolism of angiotensin I in isolated rat hearts. Effect of angiotensin converting enzyme inhibitors. Biochemical Pharmacology, 50: 1451-1459.
- 17. Neves LAA, Almeida AP, Khosla MC, Campagnole-Santos MJ & Santos RAS (1997). Effect of angiotensin-(1-7) on reperfusion arrhythmias in isolated rat hearts. Brazilian Journal of Medical and Biological Research, 30: 801-809.
- 18. Almeida AP, Fábregas BC, Madureira MM, Santos RJS, Campagnole-Santos MJ & Santos RAS (2000). Angiotensin-(1-7) potentiates the coronary vasodilatatory effect of bradykinin in the isolated rat heart. Brazilian Journal of Medical and Biological Research, 33: 709-713.
- 19. Santos RAS, Campagnole-Santos MJ, Baracho NCV, Fontes MAP, Silva LCS, Neves LAA, Oliveira DR, Caligiorne SM, Rodrigues ARV, Gropen Junior C, Carvalho WS, Simões e Silva AC & Khosla MC (1994). Characterization of a new angiotensin antagonist selective for angiotensin-(1-7): evidence that the actions of angiotensin-(1-7) are mediated by specific angiotensin receptors. Brain Research Bulletin, 35: 293-298.
- 20. Ferreira AJ, Santos RAS & Almeida AP (2001). Angiotension-(1-7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension, 38 (Part 2): 665-668.
- 21. Lubbe WF, Daries PS & Opie LH (1978). Ventricular arrhythmias associated with coronary artery occlusion and reperfusion in the isolated perfused rat heart: a model for assessment of antifibrillatory action of antiarrhythmic agents. Cardiovascular Research, 12: 212-220.
- 22. Meng H, Lonsberry BB & Pierce GN (1991). Influence of perfusate pH on the post-ischemic recovery of cardiac contractile function: involvement of sodium-hydrogen exchange. Journal of Pharmacology and Experimental Therapeutics, 258: 772-777.
- 23. Nayler WG, Poole-Wilson PA & Williams A (1979). Hypoxia and calcium. Journal of Molecular and Cellular Cardiology, 11: 683-706.
- 24. Zimmerman ANE, Daems SW, Hulsmann WC, Snijder J, Wisse E & Durrer D (1967). Morphological changes of heart muscle caused by successive perfusion with Ca free and Ca containing solutions (Ca paradox). Circulation Research, 1: 201-209.
- 25. Pabla R & Curtis MJ (1996). Effect of endogenous nitric oxide on cardiac systolic and diastolic function during ischemia and reperfusion in the rat isolated perfused heart. Journal of Molecular and Cellular Cardiology, 28: 2111-2121.
- 26. Pabla R & Curtis MJ (1995). Effect of NO modulation on cardiac arrhythmias in the rat isolated heart. Circulation Research, 77: 984-992.
- 27. Thiemermann C & Zacharowski K (2000). Selective activation of E-type prostanoid3-receptors reduces myocardial infarct size. A novel insight into the cardioprotective effects of prostaglandins. Pharmacology and Therapeutics, 87: 61-67.
- 28. Zi T, Fukui H, Nishikawa K, Hirata M & Kikuchi S (1978). Coronary vasospastic action of thromboxane A2 in isolated, working guinea pig hearts. European Journal of Pharmacology, 53: 49-56.
- 29. Coker SJ, Parratt JR, Ledingham IM & Zeitlin IJ (1981). Thromboxane and prostacyclin release from ischaemic myocardium in relation to arrhythmias. Nature, 291: 323-324.
- 30. Schmitz JM, Apprill PG, Buja LM, Willerson JT & Campbell WB (1985). Vascular prostaglandin and thromboxane production in a canine model of myocardial ischemia. Circulation Research, 57: 223-231.
- 31. Kloner RA, Ganote CE & Jennings RB (1974). The "no-reflow" phenomenon after temporary coronary occlusion in the dog. Journal of Clinical Investigation, 54: 1496-1508.
- 32. Trachte GJ, Lefer AM, Aharony D & Smith JB (1979). Potent constriction of cat coronary arteries by hydroperoxides of arachidonic acid and its blockade by anti-inflammatory agents. Prostaglandins, 18: 909-914.
- 33. Baker KM, Booz GW & Dostal DE (1992). Cardiac actions of angiotensin II. Role of an intracardiac renin-angiotensin system. Annual Review of Physiology, 54: 227-241.
- 34. Huizer T, Van der Meer P & de Jong JW (1992). Captopril inhibits angiotensin I-induced coronary flow reduction in isolated rat heart but has no effect on contractility or energy metabolism. European Heart Journal, 13: 109-114.
- 35. Ishihata A & Endoh M (1995). Species related differences in inotropic effects of angiotensin II in mammalian ventricular muscle. Receptors, subtypes and phosphoinositide hydrolysis. British Journal of Pharmacology, 114: 447-453.
- 36. Fowler NO & Holmes JC (1964). Coronary and myocardial actions of angiotensin. Circulation Research, 14: 191-201.
- 37. Traquandi C & Riva E (1998). Cardiac effects of angiotensin I and angiotensin II: dose-response studies in the isolated perfused rat heart. Pharmacological Research, 37: 57-65.
- 38. Ford WR, Clanachan AS, Robin Hiley C & Jugdutt BI (2001). Angiotensin II reduces infarct size and has no effect on post-ischaemic contractile dysfunction in isolated rat hearts. British Journal of Pharmacology, 134: 38-45.
- 39. Yoshiyama M, Kim S, Yamagishi H, Omura T, Tani T, Takagi M, Toda I, Teragaki M, Akioka K, Takeuchi K & Takeda T (1994). The deleterious effects of exogenous angiotensin I and angiotensin II on myocardial ischemia-reperfusion injury. Japanese Circulation Journal, 58: 362-368.
- 40. Loot AE, Roks AJ, Henning RH, Tio RA, Suurmeijer AJ, Boomsma F & van Gilst WH (2002). Angiotensin-(1-7) attenuates the development of heart failure after myocardial infarction in rats. Circulation, 105: 1548-1550.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
30 Aug 2002 -
Date of issue
Sept 2002
History
-
Accepted
12 Aug 2002 -
Received
03 Jan 2002
