Abstract
Abnormal riboflavin status in the absence of a dietary deficiency was detected in 31 consecutive outpatients with Parkinson's disease (PD), while the classical determinants of homocysteine levels (B6, folic acid, and B12) were usually within normal limits. In contrast, only 3 of 10 consecutive outpatients with dementia without previous stroke had abnormal riboflavin status. The data for 12 patients who did not complete 6 months of therapy or did not comply with the proposed treatment paradigm were excluded from analysis. Nineteen PD patients (8 males and 11 females, mean age ± SD = 66.2 ± 8.6 years; 3, 3, 2, 5, and 6 patients in Hoehn and Yahr stages I to V) received riboflavin orally (30 mg every 8 h) plus their usual symptomatic medications and all red meat was eliminated from their diet. After 1 month the riboflavin status of the patients was normalized from 106.4 ± 34.9 to 179.2 ± 23 ng/ml (N = 9). Motor capacity was measured by a modification of the scoring system of Hoehn and Yahr, which reports motor capacity as percent. All 19 patients who completed 6 months of treatment showed improved motor capacity during the first three months and most reached a plateau while 5/19 continued to improve in the 3- to 6-month interval. Their average motor capacity increased from 44 to 71% after 6 months, increasing significantly every month compared with their own pretreatment status (P < 0.001, Wilcoxon signed rank test). Discontinuation of riboflavin for several days did not impair motor capacity and yellowish urine was the only side effect observed. The data show that the proposed treatment improves the clinical condition of PD patients. Riboflavin-sensitive mechanisms involved in PD may include glutathione depletion, cumulative mitochondrial DNA mutations, disturbed mitochondrial protein complexes, and abnormal iron metabolism. More studies are required to identify the mechanisms involved.
Parkinson's disease; Riboflavin; Flavin-adenine dinucleotide; Glutathione; Iron; Hemin
Braz J Med Biol Res, October 2003, Volume 36(10) 1409-1417
High doses of riboflavin and the elimination of dietary red meat promote the recovery of some motor functions in Parkinson's disease patients
C.G. Coimbra1,2 and V.B.C. Junqueira3,4
1Setor de Neurologia,Hospital do Servidor Público Municipal de São Paulo, São Paulo, SP, Brasil
2Departamento de Neurologia e Neurocirurgia, Universidade Federal de São Paulo, São Paulo, SP, Brasil
3Disciplina de Geriatria, Departamento de Medicina, Centro de Estudos do Envelhecimento, Universidade Federal de São Paulo, São Paulo, SP, Brasil
4VITÆ - Cromatografia Líquida em Análises Clínicas S/C Ltda., São Paulo, SP, Brasil
Patients and Methods
Abstract
Abnormal riboflavin status in the absence of a dietary deficiency was detected in 31 consecutive outpatients with Parkinson's disease (PD), while the classical determinants of homocysteine levels (B6, folic acid, and B12) were usually within normal limits. In contrast, only 3 of 10 consecutive outpatients with dementia without previous stroke had abnormal riboflavin status. The data for 12 patients who did not complete 6 months of therapy or did not comply with the proposed treatment paradigm were excluded from analysis. Nineteen PD patients (8 males and 11 females, mean age ± SD = 66.2 ± 8.6 years; 3, 3, 2, 5, and 6 patients in Hoehn and Yahr stages I to V) received riboflavin orally (30 mg every 8 h) plus their usual symptomatic medications and all red meat was eliminated from their diet. After 1 month the riboflavin status of the patients was normalized from 106.4 ± 34.9 to 179.2 ± 23 ng/ml (N = 9). Motor capacity was measured by a modification of the scoring system of Hoehn and Yahr, which reports motor capacity as percent. All 19 patients who completed 6 months of treatment showed improved motor capacity during the first three months and most reached a plateau while 5/19 continued to improve in the 3- to 6-month interval. Their average motor capacity increased from 44 to 71% after 6 months, increasing significantly every month compared with their own pretreatment status (P < 0.001, Wilcoxon signed rank test). Discontinuation of riboflavin for several days did not impair motor capacity and yellowish urine was the only side effect observed. The data show that the proposed treatment improves the clinical condition of PD patients. Riboflavin-sensitive mechanisms involved in PD may include glutathione depletion, cumulative mitochondrial DNA mutations, disturbed mitochondrial protein complexes, and abnormal iron metabolism. More studies are required to identify the mechanisms involved.
Key words: Parkinson's disease, Riboflavin, Flavin-adenine dinucleotide, Glutathione, Iron, Hemin
Introduction
During absorption of riboflavin, flavokinase phosphorylates the vitamin to yield flavin mononucleotide (FMN) that, according to the cellular requirements, is transformed into flavin-adenine dinucleotide (FAD) by FAD synthase (1,2). Progressive deficiency of riboflavin is associated with co-factor loss in a controlled manner, apparently ensuring that essential catalytic activity such as that related to aerobic metabolism is preserved (3,4).
Low riboflavin status may also result from defective absorption. In spite of an adequate dietary intake of riboflavin (FAD, vitamin B2), 10-15% of the inhabitants of London and of Florence present low activities of two riboflavin-dependent enzymes - erythrocyte glutathione reductase (EGR) and pyridoxin(pyridoxamine)-phosphate oxidase (5). The activity of both enzymes was corrected by adding their respective co-factors (FAD or FMN) to a test tube assay or by administering high doses of riboflavin (24-30 mg per day for 5-8 weeks) to the affected individuals (6). The dependency of both FMN and FAD levels on riboflavin absorption (i.e., on flavokinase activity), and the normalization of the activities of both FMN- and FAD-dependent enzymes only at a high riboflavin intake, taken together, are consistent with the expression of flavokinase isoforms with low affinity for the substrate - riboflavin (5). Anderson et al. (5) suggested that the relatively large percent of persons with altered riboflavin absorption (10-15%) may reflect the situation in the world population rather than being a feature of a particular ethnic group.
Low EGR activity may explain glutathione depletion with impaired antioxidant defense, the earliest neurochemical abnormality in Parkinson's disease (PD), already observed in the substantia nigra before the disorder becomes clinically evident (7). Moreover, the reduced bioavailability of FMN and/or FAD may also explain the impaired oxidative metabolism of PD patients (8-10).
The first objective of the present study was to determine the status of riboflavin in PD patients. The second was to evaluate the specificity of the alterations of riboflavin status for PD by measuring the levels of vitamin B2 and of other determinants of homocystinemia (vitamins B6, B12, and folic acid) in PD patients and comparing them with those of individuals with dementia (11-13). Third, we also determined the effect of normalization of riboflavin status on the motor capacity of PD patients. Part of the data reported here, obtained during the first 3 months of treatment, were reported at the 6th International Conference on Parkinson's and Alzheimer's Diseases (14).
Patients and Methods
This study was approved by the Ethics Committee for Clinical Research of the Hospital do Servidor Público Municipal de São Paulo (HSPM) and informed consent was obtained from all participants or persons responsible for them.
The diagnosis of sporadic PD was made according to current criteria (15) with special care taken to exclude confounding disorders, particularly in the early stages of the disease.
Vitamin and homocysteine determinations were performed on 31 sporadic PD patients (67.5 ± 9.3 years old, 13 males and 18 females): 3, 3, 3, 8, and 14 patients were assigned, early in the morning, to stages I to V of Hoehn and Yahr (16), respectively.
Ten individuals (77.5 ± 8.8 years old, 5 males and 5 females) with dementia without stroke (DwoSt) and a low Mini-Mental score (13) were used as the control group for blood chemistry. They had no history of stroke or evidence for ischemic lesions of the brain by CT or NMRI and had been consecutively attended in the Neurology Clinic of HSPM.
Blood samples were obtained after a 10- to 12-h fast for serum assays of vitamin B12 by electrochemiluminescence immunoassay (11820753 Roche Diagnostics GmbH, Mannheim, Germany) and of homocysteine by HPLC (17). Heparinized plasma was assayed for FAD (18), vitamin B6 (19), and folic acid (20)by HPLC, as well as for the determination of the EGR-activation coefficient (EGR-AC) (21) in red blood cell lysates (22).
A food questionnaire covered the weekly dietary habits of all PD and DwoSt patients from 5 years prior to the onset of PD until the appearance of spontaneous changes associated with the onset of chewing and/or swallowing impairment or until the medical interview in the absence of these impairments. The questionnaire also evaluated the adequacy of daily vitamin intake.
All PD patients received 30 mg riboflavin orally at about 8-h intervals (90 mg/day) and their usual symptomatic medications. This dosage was used to avoid decreased absorption associated with higher doses or shorter intervals between administrations. Due to the renal excretion of riboflavin (3), the treatment was only initiated after confirmation of normal blood levels of creatinine (0.5-1.4 mg/dl). Because the PD patients had a higher consumption of red meat (beef and pork) than sex-matched controls (19 healthy non-consanguineous relatives or neighbors of similar age recruited for controlling the dietary habits), all PD patients were required to eliminate all red meat from their diets. The symptomatic drugs for PD in use included L-DOPA with carbidopa (200/50 mg tablets), L-DOPA with benserazide hydrochloride (200/50 mg tablets), biperiden (2 or 4 mg tablets), amantadine hydrochloride (100 mg tablets), selegiline (5 mg tablets), and pramipexole (0.25 or 1.0 mg tablets) taken alone or in diverse combinations. The treatment paradigm with symptomatic drugs for PD for each patient when the study began was maintained.
The motor capacities of the 19 PD patients who complied with the proposed treatment for 6 months by early August 2003 were rated monthly according to a motor function scale (Table 1), and compared with their own pretreatment values. The scale was based on that of Hoehn and Yahr (16) and new categories were added in order to detect subtle changes in the patients' motor capacity. In addition, the presence or absence of responses to symptomatic drugs for PD is also used for more accurate characterization of the residual motor capacity of PD patients (for instance, compare the descriptions corresponding to 0 and 15% of motor capacity, Table 1). Although there are no direct validation studies of this rating system, the different levels of motor capacity in Table 1 represent a simple increase in the number of components within stages I to V of the widely employed Hoehn and Yahr system (16).
High doses of riboflavin and the elimination of dietary red meat promote the recovery of some motor functions in Parkinson's disease patients. C.G. Coimbra and V.B.C. Junqueira. Brazilian Journal of Medical and Biological Research, 36 (10): 1409, 2003.
After the first month of treatment, compliance with the dietary directions and vitamin intake was determined in all patients, and the fasting plasma levels of FAD and EGR-AC values were re-evaluated in 9 of them approximately 9-12 h after the latest riboflavin dose.
The blood chemistry data obtained from both groups were compared statistically by the Student t-test and the motor function data were analyzed statistically by the Wilcoxon signed rank test, with the level of significance set at P < 0.05.
Results
Diversified food intake, including daily ingestion of milk, which is particularly rich in vitamin B2, was confirmed in all patients, with PD patients frequently declaring a strong preference for red meat. The content of the daily family meals was usually adapted to meet the high demand for red meat of most PD patients. In contrast, all 10 DwoSt patients passively accepted the family diet. The estimated red meat consumption prior to the onset of impaired chewing/swallowing by 19 PD patients (8 males and 11 females, mean age ± SD = 66.2 ± 8.6 years) at lunch and dinner within a 7-day period was significantly higher (mean consumption = 2,044 ± 1,439 g/week, range = 0-5,100 g/week) than that of their 19 diet controls (8 males and 11 females, all healthy individuals of similar social and cultural background, recruited among non-consanguineous relatives and neighbors of PD patients of similar age; mean age = 64.6 ± 11.3 years, mean consumption = 789 ± 509 g/week, range = 150-1800 g/week; P < 0.01, Mann-Whitney U-test). The calorie intake did not differ significantly between the two groups.
The basal plasma concentrations of FAD of the PD patients (100.9 ± 22 ng/ml) were significantly lower than those observed in the patients with DwoSt (128.8 ± 25.6 ng/ml, P < 0.01, Student t-test) while other determinants of homocysteine levels (pyridoxine, folic acid, and methylcobalamin) were usually within normal limits, and did not differ significantly between the two groups (Table 2). The PD group also had significantly higher EGR-AC levels than DwoSt patients (1.43 ± 0.26 vs 1.20 ± 0.11, respectively, P < 0.01, Student t-test).
High doses of riboflavin and the elimination of dietary red meat promote the recovery of some motor functions in Parkinson's disease patients. C.G. Coimbra and V.B.C. Junqueira. Brazilian Journal of Medical and Biological Research, 36 (10): 1409, 2003.
It is important to point out that all 31 PD patients (including 3 newly diagnosed individuals not on symptomatic drugs for PD) but only 3 of 10 DwoSt patients had low plasma riboflavin levels. Normalization of the plasma concentrations of riboflavin and EGR-AC values was confirmed after 1 month of treatment (from 106.4 ± 34.9 to 179.2 ± 23.0 ng/ml, and from 1.40 ± 0.25 to 1.11 ± 0.08, N = 9, respectively).
About 10 to 15 days after the beginning of high-dose riboflavin treatment, PD patients often reported better (progressively less interrupted) sleep at night, improved reasoning, higher motivation, and reduced depression. Their family members usually started noticing motor improvements after 20 days of treatment, but in some cases of advanced disability the patient was able to change body position in bed at night as early as on the third day of treatment.
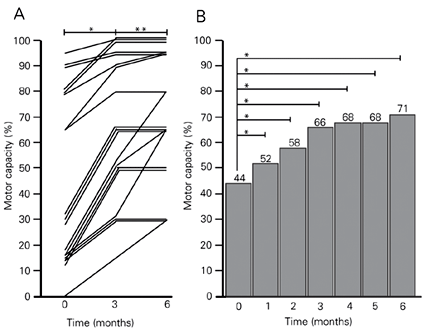
By the time of writing this report in August 2003, 19 PD patients (respectively, 3, 3, 2, 5, and 6 patients initially rated as stages I to V of Hoehn and Yahr (16)) had completed 6 months of treatment with riboflavin administration and dietary red meat elimination. The data in Figure 1A show that all of them improved their motor capacity during the first 3 months and most reached a plateau, while 5/19 continued to improve in the 3- to 6-month interval. Figure 1B shows that the average motor capacity for these 19 patients increased from 44 to 71%. Their motor capacity increased significantly during the first month and every month for the next 5 months of treatment compared with their own pretreatment status, demonstrating a progressive and marked improvement (P < 0.001, Wilcoxon signed rank test). The rate of motor recovery was higher in the first 3 months than in the last 3 months of treatment. No patient on high doses of riboflavin reported adverse effects.
Because they could stand and walk with improved (although still altered) balance by 2 months of treatment, two male patients (initially in stage V (16) with associated dementia and hallucinations) started striking imaginary persons and/or often attempted to leave home unaccompanied, reacting aggressively against the relative who tried to stop them. These episodes of agitation and aggressiveness were observed less often by the end of the third month of riboflavin treatment and disappeared thereafter, but caused transient concern and distress among their family members who initially regarded them as signs of neurological worsening.
Three patients (2 individuals initially in stage II and 1 in stage I of Hoehn and Yahr (16)) reached 100% motor capacity within the first 3 months of treatment (Figure 1A). Four patients had run out of riboflavin tablets for up to 7 days between two consecutive clinical appointments, but sustained the benefit already achieved by then.
Twelve of 31 patients initially assessed for riboflavin status who either did not complete 6 months of therapy or did not comply with the proposed treatment paradigm were excluded from statistical analysis.
Motor capacity of patients with Parkinson's disease who received 30 mg riboflavin/8 h, orally (240 mg/day) and abstained from dietary red meat for 6 months. Motor capacity was evaluated monthly for each patient by a modification of the method of Hoehn and Yahr (16) to provide a score in percent (Table 1). A, Individual data for the evolution of motor capacity of 19 patients for 0 to 3 and 3 to 6 months of treatment. *P < 0.001 for values at 3 months (month 0) compared with those before treatment; **P < 0.05 for values at 6 months compared with those obtained at 3 months (Wilcoxon signed rank test). B, The height of the columns indicates the mean motor capacity values (see Table 1) after the indicated periods of treatment. When compared with their own basal levels (month 0), highly significant and progressively higher differences were observed for each consecutive month of treatment. *P < 0.001 (Wilcoxon signed rank test).
Discussion
This study demonstrated a progressive and marked improvement of motor capacity in consecutively evaluated patients with sporadic PD who started with below normal laboratory indexes of riboflavin and who eliminated red meat from their diets while receiving high multiple daily doses of riboflavin over a period of 6 months while taking their usual symptomatic medications. The mean motor capacity of a group of 19 PD patients showed a progressive 50% recovery over a period of only 3 months - a most surprisingly high and fast improvement, considering that about 60% of nigral neurons have already been lost at the onset of manifestations of PD (15).
The initial riboflavin status was low in all 31 consecutively evaluated PD individuals, and significantly lower in PD patients compared with those with another neurodegenerative disease also associated with hyperhomocystinemia (DwoSt), suggesting that abnormal riboflavin status may be a specific feature of PD rather than a minor metabolic contributor to the degeneration of nigral neurons. Taken together with the rapid and profound neurological improvement associated with normalization of riboflavin status, this observation suggests that altered riboflavin status may be a cause of neurodegeneration in PD.
Although urinary excretion of riboflavin peaks within 1-2 h and returns to baseline within 5-6 h after a large oral dose (3), the benefit achieved did not vanish in four PD patients over a therapeutic interval of up to 7 days. This observation suggests the occurrence of steady plastic changes rather than a pharmacological effect of high-dose riboflavin treatment to account for the improved motor capacity shown in Figure 1. The steady build-up of the motor recovery observed during the first 3 months of treatment suggests that this treatment paradigm may inactivate fundamental neurodegenerative mechanisms (e.g., glutathione depletion, considered to be an early key event in the pathogenesis of PD (23,24)), possibly allowing regenerative plastic phenomena to occur.
The importance of the elimination of dietary red meat for the results reported here is not known. The content of vitamin B2 in meat in general is considerable (about 0.2 mg/100 g), and diverse cooking procedures cause only minor (7-18%) loss of this micronutrient (25). The daily requirement for individuals above the age of 14 years is £1.3 mg/day. Therefore, if the PD patients had a normal absorptive capacity for vitamin B2, their large ingestion of red meat (up to 700 g/day), associated with milk, rice and beans, fruits and vegetables, should have provided a normal riboflavin status. In contrast, 31 consecutive PD patients had laboratory evidence for riboflavin deficiency (Table 2) suggesting that patients with sporadic PD belong to the subset of the general population (10-15%) (3) that may express a flavokinase with low affinity for vitamin B2, leading to a decreased absorption.
However, the digestion of red meat releases hemin, a highly diffusible toxin that, when not properly inactivated, increases intracellular iron concentrations and enhances hydroxyl radical production (Fenton reaction). Most of the absorbed hemin is destroyed by the enzyme heme oxygenase (HO) in the digestive tract and liver (26). Because HO is oxidized during the catabolization of hemin to biliverdin, the HO molecules must be reduced through the coordinated activity of the flavoenzyme cytochrome P450 reductase for continued hemin inactivation (Figure 2) (27). Cytochrome P450 reductase is particularly sensitive to riboflavin deficiency because it requires both FMN and FAD as prosthetic groups (28). It is possible that individuals with decreased absorption of vitamin B2 may not completely inactivate high dietary levels of hemin, allowing this neurotoxic compound to reach the brain cells. Consistently, the staining for HO-1 isozyme is increased in astrocytes and reacts with neuronal Lewy bodies in the nigra of PD patients, suggesting that its overexpression may contribute to the pathological iron deposition and mitochondrial damage in PD (29). By binding glutathione (30) hemin may further decrease glutathione levels in the brains of PD patients through a direct mechanism.
Because humans lack efficient iron excretory mechanisms, iron excess is dealt with by increasing the synthesis of the iron-storage protein ferritin (31). Disturbed systemic (32) and brain (33) iron metabolism has been reported in PD, suggesting that a selective decrease in the levels of ferritin may result in an increase in intracellular free iron, thereby enhancing free radical production (34). Indeed, vitamin B2 deficiency in rodents is associated with low circulating iron concentrations, increased iron turnover and excretion into the intestinal lumen, which may occur in response to impaired ferritin synthesis (35,36). Therefore, the consistent finding of an abnormal riboflavin status in PD, as reported here, may help to explain the disturbed iron metabolism found in PD patients, with the underlying mechanisms possibly involving impaired hemin catabolism and reduced ferritin synthesis. Interestingly, the highest world prevalence of PD is found among the inhabitants of Buenos Aires (37), where the consumption of red meat is traditionally high. Similarly, the identification of high dietary animal fat as a risk factor for PD (37) may actually reflect a role of high dietary hemin in PD pathology.
Moreover, because FAD is required in the two alternative pathways of deoxynucleotide synthesis (2), DNA repair and replication are expected to be disturbed upon decreased bioavailability of riboflavin, and abnormal riboflavin status may also explain the cumulative mitochondrial DNA mutations reported in PD (38).
The present results with 19 PD patients who showed a significant improvement in motor function after treatment with riboflavin and the elimination of red meat from the diet suggest that an abnormal riboflavin status, possibly due to flavokinase deficiency, may be an essential requirement for triggering and sustaining the degeneration of dopaminergic neurons in PD. As a result of the reduced B2 bioavailability, ATP production is selectively preserved, while the less critical FAD- or FMN-dependent metabolic pathways are impaired (4). Consequently, free iron concentrations in the cytosol increase as a result of impaired ferritin synthesis and/or reduced hemin catabolism associated with hydrogen peroxide accumulation due to glutathione depletion, thereby triggering the Fenton reaction and ultimately leading to the selective formation of the potent neurotoxin 6(OH)DA in dopaminergic neurons.
Current concepts about the cause of sporadic PD suggest an inherited predisposition to environmental or endogenous toxic agents (39), and the data presented and reviewed here suggest that flavokinase deficiency should be considered in future research as a promising candidate to account for this inherited predisposition, while dietary factors such as red meat consumption may largely account for the environmental/endogenous toxicity. The administration of high doses of riboflavin combined or not with red meat elimination may be an effective therapeutic paradigm addressing the determinants of PD, capable of providing regression to earlier clinical stages, or even to the nonsymptomatic state without symptomatic drugs for PD (at least in some cases), rather than only disease stabilization or partial symptomatic relief.
Although the relentless progression of PD clearly contrasts with the results of the treatment paradigm reported here, a larger and more prolonged study is certainly required to document the steadiness and the full extent of the ongoing recovery. A scientifically desirable blinded clinical trial with a placebo would necessarily leave known riboflavin-deficient patients untreated for a long period of time, when their neurological disability may progress as a consequence of sustained loss of nigral neurons, possibly rendering the ultimate response to delayed normalization of their riboflavin levels less complete. Therefore, the need for controlled trials should be weighed ethically considering the contrast of the natural history of PD (progress of motor disability to death despite an increase in the efficacy of symptomatic drugs for PD treatment) with the outcome of the vitamin B2 treatment observed in larger and more prolonged studies without controls.
Dependency of hemin catabolism on riboflavin bioavailability. The elimination of hemin requires cyclic reduction of heme oxygenase by flavoprotein cytochrome P450 reductase that, in turn, utilizes both flavin mononucleotide (FMN) and flavin-adenine dinucleotide (FAD) as prosthetic groups. Average or increased red meat consumption may overload the capacity of this chain of reactions already compromised by impaired intestinal absorption of riboflavin (with decreased FMN and FAD synthesis), leading to increased hemin (iron) delivery to the CNS and increased utilization of riboflavin for hemin inactivation. Modified from Figure 1, box 21-1, page 783 of Ref. 2.
Acknowledgments
The authors are grateful to Mr. Terence O'Reilly (Novartis, Basel, Switzerland) for his suggestions about statistical analysis.
Address for correspondence: C.G. Coimbra, UNIFESP, Rua Pedro de Toledo, 781, 7º andar, 04039-032 São Paulo, SP, Brasil. Fax: +55-11-5539-3123. E-mail: coimbracg.nexp@epm.br
Publication supported by FAPESP. Received August 13, 2003. Accepted August 27, 2003.
- 1. Brody T (1999). Nutritional Biochemistry Academic Press, San Diego, CA, USA.
- 2. Nelson DL & Cox MM (2000). Lehninger Principles of Biochemistry Worth Publishers, New York.
- 3. Bates CJ (1997). Bioavailability of riboflavin. European Journal of Clinical Nutrition, 51: S38-S42.
- 4. Ross NS & Hansen TP (1992). Riboflavin deficiency is associated with selective preservation of critical flavoenzyme-dependent metabolic pathways. Biofactors, 3: 185-190.
- 5. Anderson BB, Scattoni M, Perry GM, Galvan P, Giuberti M, Buonocore G & Vullo C (1994). Is the flavin-deficient red blood cell common in Maremma, Italy, an important defense against malaria in this area? American Journal of Human Genetics, 55: 975-980.
- 6. Anderson BB, Perry GM, Modell CB, Child JA & Mollin DL (1979). Abnormal red-cell metabolism of pyridoxine associated with beta-thalassaemia. British Journal of Haematology, 41: 497-507.
- 7. Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ & Schapira AH (1994). Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Annals of Neurology, 35: 38-44.
- 8. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P & Marsden CD (1990). Mitochondrial complex I deficiency in Parkinson's disease. Journal of Neurochemistry, 54: 823-827.
- 9. Mytilineou C, Werner P, Molinari S, Di Rocco A, Cohen G & Yahr MD (1994). Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson's disease. Journal of Neural Transmission. Parkinson's Disease and Dementia Section, 8: 223-228.
- 10. Mizuno Y, Matuda S, Yoshino H, Mori H, Hattori N & Ikebe S (1994). An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson's disease. Annals of Neurology, 35: 204-210.
- 11. Hustad S, Ueland PM, Vollset SE, Zhang Y, Bjørke-Monsen AL & Schneede J (2000). Riboflavin as a determinant of plasma total homocysteine: effect modification by the methylenetetrahydrofolate reductase C677T polymorphism. Clinical Chemistry, 46: 1065-1071.
- 12. Diaz-Arrastia R (2000). Homocysteine and neurologic disease. Archives of Neurology, 57: 1422-1427.
- 13. Crum RM, Anthony JC, Bassett SS & Folstein MF (1993). Population-based norms for the Mini-Mental State Examination by age and educational level. Journal of the American Medical Association, 269: 2386-2391.
- 14. Coimbra CG & Junqueira VBC (2003). Altered riboflavin metabolism in Parkinson's disease: Pathophysiologic and therapeutic implications. 6th International Conference AD/PD. Alzheimer's and Parkinson's disease: new perspectives, Seville, Spain, May 8-12. Book of Abstracts, 96.
- 15. Fahn S & Przedborski S (2000). Parkinsonism. In: Rowland LP (Editor), Merrit's Neurology Lippincott Williams & Wilkins, Philadelphia, PA, USA.
- 16. Hoehn MM & Yahr MD (1967). Parkinsonism: onset, progression and mortality. Neurology, 17: 427-442.
- 17. Pfeiffer CM, Huff DL & Gunter EW (1999). Rapid and accurate HPLC assay for plasma total homocysteine and cysteine in a clinical laboratory setting. Clinical Chemistry, 45: 290-292.
- 18. Speek AJ, van Schaik F, Schrijver J & Schreurs WH (1982). Determination of the B2 vitamer flavin-adenine dinucleotide in whole blood by high-performance liquid chromatography with fluorometric detection. Journal of Chromatography, 228: 311-316.
- 19. Sharma SK & Dakshinamurti K (1992). Determination of vitamin B6 vitamers and pyridoxic acid in biological samples. Journal of Chromatography, 578: 45-51.
- 20. Kelly P, McPartlin J & Scott J (1996). A combined high-performance liquid chromatographic-microbiological assay for serum folic acid. Analytical Biochemistry, 238: 179-183.
- 21. Sauberlich HE, Judd JH, Nichoalds GE, Broquist HP & Darby WJ (1972). Application of the erythrocyte glutathione reductase assay in evaluating riboflavin nutritional status in a high school student population. American Journal of Clinical Nutrition, 25: 756-762.
- 22. Beutler E (1975). The preparation of red cells for assay. In: Beutler E (Editor), Red Cell Metabolism. A Manual of Biochemical Methods 2nd edn. Grune and Straton, New York.
- 23. Jenner P, Dexter DT, Sian J, Shapira AHV & Marsden CD (1992). Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. Annals of Neurology, 32: S82-S87.
- 24. Schulz JB, Lindenau J, Seyfried J & Dichgans J (2000). Glutathione, oxidative stress and neurodegeneration. European Journal of Biochemistry, 267: 4904-4911.
- 25. Pinheiro-Sant'ana HM, Stringueta PC & Penteado MDVC (1999). Stability of B-vitamins in meats prepared by foodservice. 2. Riboflavin. Foodservice Research International, 11: 53-67.
- 26. Brown EB, Hwang Y-F, Nichol S & Ternberg J (1968). Absorption of radioiron-labeled hemoglobin by dogs. Journal of Laboratory and Clinical Medicine, 72: 58-64.
- 27. Ryter SW & Tyrrel RM (2000). The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radical Biology and Medicine, 28: 289-309.
- 28. Wang M, Roberts DL, Paschke R, Shea TM, Masters BSS & Kim J-JP (1997). Three-dimensional structure of NADPH-cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proceedings of National Academy of Sciences, USA, 94: 8411-8416.
- 29. Shipper HM (2000). Heme oxygenase-1: role in brain aging and neurodegeneration. Experimental Gerontology, 35: 821-830.
- 30. Sahini VE, Dumitrescu M, Volanschi E, Birla L & Diaconu C (1966). Spectral interferometrical study of the interaction of hemin with glutathione. Biophysical Chemistry, 58: 245-253.
- 31. Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA & Klausner RD (1988). Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science, 240: 924-928.
- 32. Logroscino G, Marder K, Graziano J, Freyer G, Slavkovich V, LoIacono N, Cote L & Mayeux R (1997). Altered systemic iron metabolism in Parkinson's disease. Neurology, 49: 714-717.
- 33. Dexter DT, Carayon A, Vidailhet M, Ruberg M, Agid F, Agid Y, Lees AJ, Wells FR, Jenner P & Marsden CD (1990). Decreased ferritin levels in brain in Parkinson's disease. Journal of Neurochemistry, 55: 16-20.
- 34. Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD & Schapira AH (1994). Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Annals of Neurology, 36: 876-881.
- 35. Adelekan DA & Thurnham DI (1986). A longitudinal study of the effect of riboflavin status on aspects of iron storage in the liver of growing rats. British Journal of Nutrition, 56: 171-179.
- 36. Powers HJ, Weaver LT, Austin S, Wright AJ & Fairweather-Tait SJ (1991). Riboflavin deficiency in the rat: effects on iron utilization and loss. British Journal of Nutrition, 65: 487-496.
- 37. Tanner CM, Goldman SM & Ross GW (2002). Etiology of Parkinson's disease. In: Jankovik JJ & Tolosa E (Editors), Parkinson's Disease and Movement Disorders Lippincott Williams & Wilkins, Philadelphia, PA, USA.
- 38. Di Monte DA (1991). Mitochondrial DNA and Parkinson's disease. Neurology, 41: 38-42.
- 39. Jenner P, Shapira AH & Marsden CD (1992). New insights into the cause of Parkinson's disease. Neurology, 42: 2241-2250.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
16 Sept 2003 -
Date of issue
Oct 2003
History
-
Accepted
27 Aug 2003 -
Received
13 Aug 2003