Abstract
The present study focused on the role of sympathetic renal nerve activity, in mediating congestive heart failure-induced sodium retention following experimental chronic myocardial infarction. Groups of male Wistar rats (240-260 g) were studied: sham-operated coronary ligation (CON3W, N = 11), coronary ligation and sham-operated renal denervation (INF3W, N = 19), 3 weeks of coronary ligation and sympathetic renal nerve denervation (INF3WDX, N = 6), sham-operated coronary ligation (N = 7), and 16 weeks of coronary ligation (INF16W, N = 7). An acute experimental protocol was used in which the volume overload (VO; 5% of body weight) was applied for 30 min after the equilibration period of continuous iv infusion of saline. Compared to control levels, VO produced an increase (P < 0.01, ANOVA) in urine flow rate (UFR; 570%) and urinary sodium excretion (USE; 1117%) in CON3W. VO induced a smaller increase (P < 0.01) in USE (684%) in INF3W. A similar response was also observed in INF16W. In INF3WDX, VO produced an immediate and large increase (P < 0.01) in UFR (547%) and USE (1211%). Similarly, in INF3W VO increased (P < 0.01) UFR (394%) and USE (894%). Compared with INF3W, VO induced a higher (P < 0.01) USE in INF3WDX, whose values were similar to those for CON3W. These results suggest that renal sympathetic activity may be involved in sodium retention induced by congestive heart failure. This premise is supported by the observation that in bilaterally renal denervated INF3WDX rats myocardial infarction was unable to reduce volume expansion-induced natriuresis. However, the mechanism involved in urinary volume regulation seems to be insensitive to the factors that alter natriuresis.
Renal denervation; Heart failure; Renal sympathetic nerves; Renal excretory function; Urinary sodium excretion
Braz J Med Biol Res, February 2004, Volume 37(2) 285-293
Chronic experimental myocardial infarction produces antinatriuresis by a renal nerve-dependent mechanism
D.R.B. Souza, J.G. Mill and A.M. Cabral
Departamento de Ciências Fisiológicas, Centro Biomédico, Universidade Federal do Espírito Santo, Vitória, ES, Brasil
References
Abstract
The present study focused on the role of sympathetic renal nerve activity, in mediating congestive heart failure-induced sodium retention following experimental chronic myocardial infarction. Groups of male Wistar rats (240-260 g) were studied: sham-operated coronary ligation (CON3W, N = 11), coronary ligation and sham-operated renal denervation (INF3W, N = 19), 3 weeks of coronary ligation and sympathetic renal nerve denervation (INF3WDX, N = 6), sham-operated coronary ligation (N = 7), and 16 weeks of coronary ligation (INF16W, N = 7). An acute experimental protocol was used in which the volume overload (VO; 5% of body weight) was applied for 30 min after the equilibration period of continuous iv infusion of saline. Compared to control levels, VO produced an increase (P < 0.01, ANOVA) in urine flow rate (UFR; 570%) and urinary sodium excretion (USE; 1117%) in CON3W. VO induced a smaller increase (P < 0.01) in USE (684%) in INF3W. A similar response was also observed in INF16W. In INF3WDX, VO produced an immediate and large increase (P < 0.01) in UFR (547%) and USE (1211%). Similarly, in INF3W VO increased (P < 0.01) UFR (394%) and USE (894%). Compared with INF3W, VO induced a higher (P < 0.01) USE in INF3WDX, whose values were similar to those for CON3W. These results suggest that renal sympathetic activity may be involved in sodium retention induced by congestive heart failure. This premise is supported by the observation that in bilaterally renal denervated INF3WDX rats myocardial infarction was unable to reduce volume expansion-induced natriuresis. However, the mechanism involved in urinary volume regulation seems to be insensitive to the factors that alter natriuresis.
Key words: Renal denervation, Heart failure, Renal sympathetic nerves, Renal excretory function, Urinary sodium excretion
Introduction
Renal sodium retention is a common characteristic of diseases such as nephrotic syndrome, hepatic cirrhosis, some experimental hypertension models, and heart failure. Sodium retention in heart failure is due to alterations in several volume control mechanisms such as the renin-angiotensin system (1,2), prostaglandins (3,4), natriuretic peptides (2,4,5), vasopressin (6), and the sympathetic nervous system (2,5,7,8). The activation of sodium-retention pathways may be viewed as a compensatory mechanism elicited to preserve the hemodynamic balance despite the reduced cardiac output. Activation of the sympathetic nervous system seems to be one of the more important mechanisms leading to sodium retention since the increase in the sympathetic outflow to the kidneys produces an important increase of sodium and water reabsorption throughout the entire nephron (9,10). Moreover, sympathetic activation may also induce sodium retention by increasing renin production and by modulating the resistance of afferent glomerular arterioles (11).
Left ventricular dysfunction produced by myocardial infarction is a well-known model of heart failure, which can be identified by the increase of left ventricular end-diastolic pressure, and the decrease of dP/dt max, stroke volume and arterial blood pressure (12). The depression of left ventricular performance in infarcted hearts can be detected by diverse sensing mechanisms, which modulate efferent pathways that interfere with renal hemodynamics and with several tubular ion and fluid transport systems (13,14). The increased sympathetic drive directed at the kidneys is one of these efferent pathways activated in heart failure (9,10).
The purpose of the present study was to investigate the participation of sympathetic neurons in the natriuretic response elicited in rats with chronic myocardial infarction. Thus, we examined whether the reduced sodium excretion observed in infarcted animals was due to changes in renal hemodynamics induced by heart failure and/or involving renal sympathetic nerve activity.
Material and Methods
Experiments were performed on male Wistar rats (240-260 g) obtained from our Department facility. All procedures were conducted in accordance with the biomedical research guidelines for the care and use of laboratory animal as stated by the Federation of Brazilian Societies of Experimental Biology.
Rats were housed in groups in a temperature- and humidity-controlled room with a 12-h light/dark cycle. Standard rat chow (Na+ content 163 mEq/kg) and tap water were available ad libitum. Five groups of rats were studied. Sham-operated coronary ligation (CON3W, N = 11) rats studied 3 weeks after sham coronary artery ligation; coronary ligation and sham-operated renal denervation (INF3W, N = 19) rats studied 3 weeks after myocardial infarction; INF3WDX (N = 6) rats studied 3 weeks after sympathetic renal denervation and coronary artery ligation; CON16W (N = 7) rats studied 16 weeks after sham surgery for coronary ligation; INF16W (N = 7) rats studied 16 weeks after myocardial infarction.
Surgical procedures
Myocardial infarction. Coronary artery ligation was performed under ether anesthesia as previously described (15). Briefly, through a thoracotomy performed at the fourth left intercostal space, the heart was rapidly exposed and the descending anterior branches of the left coronary artery were ligated between the tip of the left atrial appendage and the pulmonary artery outflow tract with 6-0 mononylon thread. The heart was then returned to its original position in the thorax, and the thoracotomy was closed. Most animals recovered spontaneous respiratory movements after the thorax was closed. A brief period of positive ventilation was used in cases of respiratory arrest after thoracotomy closure. The mortality rate was about 20% and most of the cases occurred during the first 15 min after surgery. Control rats underwent sham surgery in which all surgical steps were performed except that the suture around the coronary vessels was not tied.
Bilateral renal denervation
Bilateral renal denervation was performed in order to eliminate the influence of renal nerves on renal excretory function. For this purpose, rats underwent chronic bilateral renal denervation 3 days before the surgical procedures for myocardial infarction. Under ketamine (30 mg/kg, im) and xylazine (3 mg/kg, im) anesthesia, the left kidney was exposed via a flank incision. The adventitia surrounding the renal artery and vein was stripped and all visible renal nerves were cut under a microscope (D.F. Vasconcellos 902, São Paulo, SP, Brazil). The vessels were then treated with 95% alcohol containing 10% phenol. After renal denervation the flank incision was sutured and the procedure repeated on the opposite side to denervate the right kidney. This renal denervation procedure prevents the renal vasoconstrictor response to suprarenal lumbar sympathetic nerve stimulation, prevents the antinatriuretic response to environmental stress and reduces renal tissue norepinephrine concentration to <5% of control for up to 15 days post-denervation (16). After the acute experiments the kidneys were kept frozen to measure norepinephrine concentration. The renal denervation procedure reduced renal tissue norepinephrine concentration to undetectable values (the control value was 424 ± 27 ng/g), removing the influence of renal nerves on kidney function.
Catheter implantation. On the day of the acute experiment, rats were anesthetized with 50 mg/kg sodium thiopental, ip, supplemented iv as needed (Cristalia, São Paulo, SP, Brazil). Catheters (PE-50 connected to PE-10) were placed into the femoral artery for recording arterial blood pressure and into the femoral vein for administration of drugs or isotonic saline infusion. According to the standard procedure used in our laboratory, the catheters were tunneled subcutaneously to the back of the neck, flushed and plugged. For urinary measurements, a suprapubic incision was then made and a bladder catheter (flanged PE-240) was inserted and sutured into the urinary bladder. The bladder catheter was then exteriorized and secured by suturing it to adjacent muscle and skin. After all the maneuvers to assess excretory renal function and after the execution of the acute protocol, a new catheter (PE-50) was placed into the left ventricle via the right carotid artery to measure systolic pressure and end diastolic pressure.
Experimental protocols
Renal excretory responses elicited by volume expansion in conscious rats. Experiments were performed on conscious rats to compare the natriuretic and diuretic responses induced by water and salt overload. Before volume expansion, the animals were submitted to iv infusion of isotonic saline (55 µl/min) to enhance the renal excretion of water and sodium, and these levels tended to stabilize and remain constant approximately 120 min after the beginning of infusion (17). Therefore, after equilibration and stabilization of the renal excretory responses, three consecutive control urine samples (C1, C2 and C3) were collected (10 min each). Urine was then collected during 3 consecutive 10-min experimental periods (E1, E2 and E3) with volume overload (5% body weight of isotonic saline administered iv over a period of 30 min). After volume overload, a 30-min recovery period consisting of three consecutive 10-min urine collection periods (R1, R2 and R3) was studied. Urinary volume and sodium content, mean arterial pressure and heart rate were determined in the above protocol, which was applied to all groups. For saline infusion, we used an infusion pump, model 600-900V (Harvard Apparatus, Dover, MA, USA). The arterial catheter was connected to a pressure transducer, model P23Db (Statham, Oxnard, CA, USA). Throughout the experiment mean arterial pressure and heart rate were continuously recorded using a polygraph (Sensormedics Dynograf Recorder R 711, Anaheim, CA, USA). During surgery and the experimental protocol the body temperature of rats was maintained at 37 ± 1ºC with a heat lamp.
At the end of the experimental protocol, the rat was anesthetized with urethane (1.5 g/kg, iv) and the right carotid artery was dissected. A PE-50 catheter was introduced and advanced into the left ventricular cavity to measure peak systolic and end-diastolic pressures. After these measurements the rat was exsanguinated and the lungs, kidneys and liver were removed, blotted and weighed.
Determination of infarct size
At the end of each experiment (renal and hemodynamic measurements), the heart was rapidly removed and perfused for 5 min with oxygenated Krebs-Henseleit solution through the aortic stump. After blood washout, the ventricular chambers were dissected, blotted, and weighed. The presence of infarction was easily confirmed by gross visualization of the fibrous scar on the anterolateral region of the left ventricle. The scar tissue was separated from the remaining left ventricular myocardium in the infarcted hearts under a microscope. Both fragments were blotted and weighed, and their outlines were drawn on graph paper to estimate the respective areas. Infarct size was calculated and reported as percent of the left ventricular endocardial surface covered with scar tissue (18). Only hearts with an infarct covering 20 to 40% of the left ventricular surface were included in this study.
Other methods
Changes in mean arterial pressure and heart rate produced by drug administration were calculated directly from the polygraph records. The kidneys were removed, rinsed in physiological saline, decapsulated, blotted and weighed for normalization of renal excretory data. Urine volume was determined gravimetrically. Urine sodium concentration was measured by flame photometry with a Micronal apparatus, model B (São Paulo, SP, Brazil). Urine and plasma inulin and p-amino-hippurate (PAH) concentrations were determined by the anthrone (19) and ethylenediamine (20) methods, respectively. Glomerular filtration rate (GFR) was measured as inulin clearance (CIN) = (VUIN)/PIN, where UIN and PIN are the urine and plasma inulin concentrations, respectively, and V is the urine flow rate. Effective renal plasma flow was determined by PAH clearance (CPAH) = (VUPAH)/PPAH, where UPAH and PPAH are the urine and plasma PAH concentrations, respectively.
Statistical analysis
All data are reported as means ± SEM. Data were analyzed statistically by repeated measures analysis of variance for the main effects and interactions and by the Tukey test for pairwise comparisons among the means. Statistical significance was set at P < 0.05.
Results
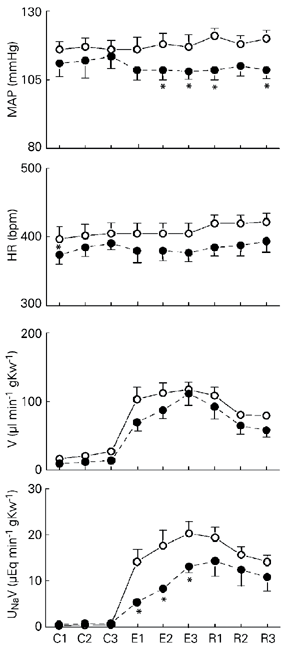
Figure 1 shows the systemic cardiovascular and renal excretory responses produced by volume overload in the intact (CON3W) and 3-week-infarcted (INF3W) rats. Throughout the experimental protocol, conscious rats received a continuous infusion (55 µl/min) of 0.9% NaCl. Mean (± SEM) data are reported for each cardiovascular and renal excretory parameter during 3 consecutive 10-min control periods (C1, C2 and C3), 3 consecutive 10-min volume overload periods, and 3 consecutive 10-min recovery periods. When compared to control levels (Figure 1, C1-C3), volume overload produced an immediate and profound increase in urine flow rate in CON3W rats (570%; from C3 = 23.7 ± 5.2 µl min-1 g kidney weight-1 (gKw-1) at 20 min, to the second experimental period (E2) = 137 ± 21 µl min-1 gKw-1) and urinary sodium excretion (1117%; from C3 = 2.05 ± 0.7 to E2 = 22.9 ± 4.1 µEq min-1 gKw-1).
Similarly, the volume expansion in INF3W rats (Figure 1), significantly (P < 0.01) increased urine flow rate compared to the pre-overload volume control level (473%; from C3 = 23.4 ± 4.5 to E2 = 110.6 ± 11 µl min-1 gKw-1), and urinary sodium excretion (684%, from C3 = 2.09 ± 0.5 µEq min-1
gKw-1, to E2 = 14.3 ± 0.5; Figure 1). Volume overload induced a significantly higher (P < 0.01) increase in urinary sodium excretion in CON3W rats. In contrast, the volume overload-induced increase in urine flow rate did not differ between intact and infarcted rats at any time period. The volume overload failed to alter any cardiovascular parameter in either the CON3W (115 ± 4 mmHg and 403 ± 12 bpm) or INF3W (110 ± 3 mmHg and 363 ± 7 bpm) group (Figure 1). The reduced levels of mean arterial pressure and heart rate are consistent with the reduction in systolic pressures observed in INF3W when compared to intact CON3W rats (Table 1).
Chronic experimental myocardial infarction produces antinatriuresis by a renal nerve-dependent mechanism. D.R.B. Souza, J.G. Mill and A.M. Cabral. Brazilian Journal of Medical and Biological Research, 37 (2): 285, 2004.
Figure 2 shows the systemic cardiovascular and renal excretory responses produced by volume overload in the 3-week-infarcted plus chronic bilaterally renal denervated (INF3WDX) and INF3W rats. Conscious rats were submitted to the same experimental protocol as described in Figure 1. Compared to control levels (Figure 2, C3), volume overload produced an immediate and profound increase in urine flow rate in INF3WDX rats (547%, from C3 = 23 ± 5.9 to E2 = 126 ± 9 µl min-1 gKw-1) and urinary sodium excretion (1211%, from C3 = 1.8 ± 0.5 to E2 = 21.8 ± 1.8 µEq min-1 gKw-1) during the second experimental period.
Similarly, volume overload in 3-week-infarcted rats without sympathetic renal denervation (INF3W) (Figure 2) significantly (P < 0.01) increased urine flow compared to the pre-overload volume control level (394%, from C3 = 27.6 ± 9.6 to E2 = 109 ± 16 µl min-1 gKw-1) and urinary sodium excretion (894%, from C3 = 1.6 ± 0.3 to E2 = 14.3 ± 2 µEq min-1 gKw-1). Compared with 3-week INF3W rats (Figure 2), volume overload induced a significantly higher (P < 0.01) increase in urinary sodium excretion in INF3WDX rats, whose values did not differ from those observed in intact control rats (Figure 1, CON3W). As observed between CON3W and INF3W, the volume overload-induced increase in urine flow rate did not differ between INF3W and INF3WDX rats at any time period. Also, volume expansion failed to alter any cardiovascular parameter in either the INF3W (104 ± 4 mmHg and 405 ± 10 bpm) or INF3WDX (111 ± 5 mmHg and 440 ± 15 bpm) group (Figure 2).
Figure 3 shows the systemic cardiovascular and renal excretory responses produced by volume overload in intact (CON16W) and chronic 16-week infarcted (INF16W) rats. Conscious rats were submitted to the same experimental protocol as described in Figure 1. Compared to control levels (Figure 3, C3), volume overload applied to CON16W rats produced an immediate and profound increase in urine flow rate (414%, from C3 = 27 ± 7.1 to E2 = 112 ± 15 µl min-1 gKw-1) and urinary sodium excretion (2514%, from C3 = 0.7 ± 0.07 to E2 = 17.6 ± 3.3 µEq min-1 gKw-1) by the second experimental period.
Similarly, volume overload applied to INF16W rats (Figure 3) significantly (P < 0.01) increased urine flow compared to the pre-overload volume control level (436%, from C3 = 20 ± 5.3 to E2 = 87.3 ± 12 µl min-1 gKw-1) and urinary sodium excretion (1037%, from C3 = 0.8 ± 0.3 to E2 = 8.3 ± 0.7 µEq min-1 gKw-1). Compared with INF16W rats, volume overload induced a significantly higher (P < 0.01) increase in urinary sodium excretion in CON16W rats. In contrast, the volume overload-induced increase in urine flow rate did not differ between intact and infarcted rats, at any time period. Volume overload failed to alter any cardiovascular parameter in either the CON16W (116 ± 4 mmHg and 405 ± 16 bpm) or INF3W (113 ± 4 mmHg and 391 ± 10 bpm) group (Figure 3).
There were no significant changes in GFR before and/or during volume overload in either CON3W (C3 = 1.25 ± 0.2 ml min-1 gKw-1) or INF3W (C3 = 1.20 ± 0.1 ml min-1 gKw-1) rats. However, effective renal plasma flow was increased (P < 0.05) from 2.5 ± 0.4 ml min-1 gKw-1 (C3) to 5.8 ± 0.9 ml min-1 gKw-1 (E2) by volume overload in CON3W rats. This significant (P < 0.05) increase induced by volume overload did not differ between INF3W (C3 = 3.9 ± 0.6; E2 = 7.4 ± 1.2 ml min-1 gKw-1) and CON3W rats.
Quantitative aspects of the evolution of myocardial infarcts. The manipulation of sham-operated rats produced no myocardial necrosis and no asymmetry in the ventricular wall thickness. The infarcted area of INF3W rats involved 36 ± 3% of the total surface area of the left ventricle (Table 1). Previous sympathetic renal denervation did not change significantly the onset and development of surface area of the infarcted wall (32 ± 3%) in INF3WDX rats (Table 1). In INF16W, the surface area of the infarcted wall involved 36 ± 1% of the total left ventricle area (Table 1).
Effect of volume overload in conscious 3-week-sham-operated coronary ligation (CON3W, open circles, N = 11) and 3-week-coronary ligation (INF3W, filled circles, N = 13) rats. Volume overload was obtained with 0.9% NaCl iv corresponding to 5% body weight. Data are reported as means ± SEM, showing the cardiovascular and renal responses during the control (C1, C2, C3), volume overload (E1, E2, E3), and recovery (R1, R2, R3) periods. During the control and recovery periods the animals were infused with isotonic saline at 55 µl/min. gKw = g kidney weight; HR = heart rate; MAP = mean arterial pressure; UNaV = urinary sodium excretion; V = urine flow rate. *P < 0.01 vs sham-operated rats (ANOVA followed by Tukey test).
Effect of volume overload in conscious 3-week-coronary ligation and sham-operated renal denervation (INF3W, filled circles, N = 6) and 3-week-sympathetic renal denervation plus coronary artery ligation (INF3WDX, open circles, N = 6) rats. Volume overload was obtained with 0.9% NaCl iv corresponding to 5% body weight. Data are reported as means ± SEM, showing the cardiovascular and renal responses during the control (C1, C2, C3), volume overload (E1, E2, E3), and recovery (R1, R2, R3) periods. During the control and recovery periods the animals were infused with isotonic saline at 55 µl/min. gKw = g kidney weight; HR = heart rate; MAP = mean arterial pressure; UNaV = urinary sodium excretion; V = urine flow rate. *P < 0.01 vs sham rats (ANOVA followed by Tukey test).
Effect of volume overload (0.9% NaCl iv 5% body weight) in conscious 16-week-sham-coronary ligation (CON16W, open circles, N = 7) and 16-week-coronary ligation (INF16W, filled circles, N = 7) rats. Volume overload was obtained with 0.9% NaCl iv, corresponding to 5% body weight. Data are reported as means ± SEM, showing the cardiovascular and renal responses during the control (C1, C2, C3), volume overload (E1, E2, E3), and recovery (R1, R2, R3) periods. During the control and recovery periods the animals were infused with isotonic saline at 55 µl/min. gKw = g kidney weight; HR = heart rate; MAP = mean arterial pressure; UNaV = urinary sodium excretion; V = urine flow rate. *P < 0.01 vs sham rats (ANOVA followed by Tukey test).
Discussion
The purpose of the present study was to examine the role of sympathetic renal nerve activity in mediating congestive heart failure-induced sodium retention following experimental myocardial infarction. An experimental protocol was performed in which the animals were submitted to a volume overload (5% of body weight) over 30 min by continuous iv infusion of isotonic saline. This volume overload produced a profound and immediate increase in urine flow rate and urinary sodium excretion. The results of the present study showed that 3 and 16 weeks of myocardial infarction significantly reduced sodium excretion during this volume expansion. The renal sympathetic nerves seem to be involved in this process since impairment of volume overload-induced natriuresis was significantly attenuated in the rats with a 3-week-old myocardial infarction previously submitted to bilateral sympathetic renal denervation, as observed in Figure 2. In view of previous findings (7), our data suggest that the ability of intravenous saline loading to induce natriuresis is impaired in conscious rats 21 days after left coronary artery ligation. In contrast, however, the cited investigators (7) observed a similar impairment in urine flow rate in these infarcted rats of the same percent magnitude as that observed in sodium excretion in renal denervated animals. In contrast to our results, DiBona (9) and Kopp and DiBona (10), in experimental models of edema formation such as myocardial infarction-induced heart failure, showed that the activation of efferent renal sympathetic nerve activity elicits sodium and water retention via renal renin secretion rate, an increase in net renal tubular sodium and water reabsorption rate, and a decrease in GFR and renal blood flow. Although we have no evidence concerning renin secretion, our data show that changes in GFR and renal blood flow do not seem to be involved in sodium retention since we did not observe difference in GFR before or during volume overload between control and infarcted groups. Similarly, heart failure (in INF3W rats) did not change the increase in renal plasma flow induced by volume overload compared to control (CON3W) animals. These dissociated effects of sympathetic activity on renal excretory function could be explained by diverse levels of efferent renal sympathetic activity elicited by a different frequency range, in agreement with studies indicating that under certain conditions the stimulus is generally too low to influence renal hemodynamics (21).
Left coronary artery ligation in the rat provides an animal model of myocardial infarction in which a wide range of infarct sizes and left ventricular dysfunction can be produced (15,22). Our data show that the size of the infarcted area in both the INF3W and INF16W groups, whose values did not differ from one another agree with those reported in literature. In both groups the left ventricular end-diastolic pressure was significantly increased (P < 0.01), characterizing ventricular dysfunction. In addition to these changes, an increase in sympathetic activity seems to be an important regulatory alteration. Increased activity of the sympathetic nervous system has been implicated in the morbidity induced by myocardial infarction (23,24), and this augmented sympathetic activity facilitates the development of cardiac arrhythmias and the increased extension of the ischemic injury (25) and contributes to some extracardiac regulatory alterations such as sodium balance, with sodium retention expanding extracellular fluid volume (14). This compensatory mechanism may be seen as a compensatory phenomenon acting to preserve the hemodynamic balance despite the decreased cardiac output induced by ventricular dysfunction. Regarding sodium retention, the involvement of renal sympathetic nerves seems to be one of the most important mechanisms because the increase of the sympathetic drive to the kidneys potentiates the renal tubular sodium and water reabsorption (9,10). Moreover the sympathetic discharge also determines sodium retention by interacting with the renin-angiotensin system and afferent artery resistance (11).
Our observations suggest that, under heart failure, the renal sympathetic nerves may participate in mediating an impaired level of sodium excretion by activating a neural sympathetic activity that influences the net renal tubular sodium reabsorption. However, the mechanism involved in urinary volume regulation seems to be insensitive to the factors that influence natriuresis. These findings serve to illustrate the importance of a neural pathway of sodium excretion (inhibitory) that seems to be potentiated after myocardial infarction. Therefore, the sympathetic activity directed at the kidneys may be a key factor contributing to the pathophysiology of heart failure development after myocardial infarction.
It may be argued that the reduced sodium excretion observed in infarcted animals was due to changes in renal hemodynamics induced by heart failure. However, the GFR and the saline loading-induced increase in effective renal plasma flow were not altered by myocardial infarction, thus excluding the possibility that changes in renal hemodynamics contributed to the depressed renal excretory response. Taken together, these data indicate that the activity of sympathetic renal nerves on tubular sodium reabsorption seems to be a leading mechanism for the reduction of sodium excretion observed here in infarcted rats.
Address for correspondence: A.M. Cabral, Programa de Pós-Graduação em Ciências Fisiológicas, Centro Biomédico, UFES, Av. Marechal Campos, 1468, 29040-090 Vitória, ES, Brasil. Fax: +55-27-335-7330. E-mail: acabral@npd.ufes.br
Research supported by CNPq and CAPES. Received February 25, 2002. Accepted October 14, 2003.
- 1. Eiskjaer H, Bagger JP, Danielsen H, Jensen JD, Jespersen B, Thomsen K, Sorensen SS & Pedersen EB (1991). Mechanisms of sodium retention in heart failure: relation to the renin-angiotensin-aldosterone system. American Journal of Physiology, 260: F883-F889.
- 2. Redfield MM, Aarhus LL, Scott Wright R & Burnett JC (1993). Cardiorenal and neurohumoral function in a canine model of early left ventricular dysfunction. Circulation, 87: 2016-2022.
- 3. Criado M, Flores O, Hidalgo F, Lopez-Novoa JM & Sanchez-Rodriguez A (1999). Interaction between prostanoids and nitric oxide in the control of tubular function in rats with chronic bile ligation. Canadian Journal of Pharmacology, 77: 111-117.
- 4. Villarreal D, Freeman RH, Habibullah AA & Simmons JC (1997). Indomethacin attenuates the renal actions of atrial natriuretic factor in dogs with chronic heart failure. American Journal of Medical Sciences, 314: 67-72.
- 5. Koepke JP & DiBona GF (1987). Blunted natriuresis to atrial natriuretic peptide in chronic sodium-retaining disorders. American Journal of Physiology, 252: F865-F871.
- 6. Wong LL & Verbalis JG (2001). Vasopressin V2 receptor antagonists. Cardiovascular Research, 51: 391-402.
- 7. DiBona GF, Herman PJ & Sawin LL (1988). Neural control of renal function in edema-forming states. American Journal of Physiology, 254: R1017-R1024.
- 8. DiBona GF & Sawin LL (1991). Role of renal nerves in sodium retention of cirrhosis and congestive heart failure. American Journal of Physiology, 260: R298-R305.
- 9. DiBona GF (1985). Neural control of renal function. Federation Proceedings, 44: 2815-2850.
- 10. Kopp UC & DiBona GF (1987). Neural control of volume homeostasis. In: Brenner BM & Stein JH (Editors), Body Fluid Homeostasis. Contemporary Issues in Nephrology Vol. 16. Churchill Livingstone, New York, 185-220.
- 11. Ammons WS, Koyama S & Manning JW (1982). Neural and vascular interaction in renin response to graded renal nerve stimulation. American Journal of Physiology, 242: R552-R562.
- 12. Swan HJC, Forrester JS, Diamond G, Chatterjee K & Parmley WW (1972). Hemodynamic spectrum of myocardial infarction and cardiogenic shock. Circulation, 45: 1097-1110.
- 13. Humes DH, Gottlieb MN & Brenner BM (1978). The kidney in congestive heart failure. In: Brenner BM & Stein JS (Editors), Contemporary Issues in Nephrology: Sodium and Water Homeostasis Vol. 2. Churchill Livingstone, New York, 51-72.
- 14. Skorecki KL & Brenner BM (1982). Body fluid homeostasis in congestive heart failure and cirrhosis with ascites. American Journal of Medicine, 72: 323-338.
- 15. Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA & Braunwald E (1979). Myocardial infarct size and ventricular function in rats. Circulation Research, 44: 503-512.
- 16. DiBona GF & Sawin LL (1983). Renal nerves in renal adaptation to dietary sodium restriction. American Journal of Physiology, 245: F322-F328.
- 17. Cabral AM, Varner KJ & Kapusta DR (1997). Renal excretory responses produced by central administration of opioid agonists in ketamine and xylazine-anesthetized rats. Journal of Pharmacology and Experimental Therapeutics, 282: 609-616.
- 18. Mill JG, Stefanon I, Leite CM & Vassallo DV (1990). Changes in performance of the surviving myocardium after left ventricular infarction in rats. Cardiovascular Research, 24: 748-753.
- 19. Fuhr J, Veelken R, Becker H, Ganten D, Lang RE & Unger T (1986). Eine einfache colorimetrische Methode zur Inulinbestimmung fur Nierenclearance Untersuchungen bei Stoffwechselgesunden und Diabetikern. Klinische Wochenschrift, 33: 729-730.
- 20. Bratton AC & Marshall EK (1939). A new coupling component for sulfanilamide determination. Journal of Biological Chemistry, 128: 537-550.
- 21. DiBona GF & Kopp UC (1997). Neural control of renal function. Physiological Reviews, 77: 75-197.
- 22. Fletcher PJ, Pfeffer JM, Pfeffer MA & Braunwald E (1981). Left ventricular diastolic pressure-volume relations in rats with healed myocardial infarction. Effects on systolic function. Circulation Research, 49: 618-626.
- 23. Ceremuzynski L, Staszewska-Barczak J & Herbaczynska-Cedro K (1969). Cardiac rhythm disturbance and the release of catecholamines after acute coronary occlusion in dogs. Cardiovascular Research, 3: 190-197.
- 24. Coor PB, Pearle DL, Hilton JR, Roberts WC & Gillis RA (1976). Site of myocardial infarction. A determinant of the cardiovascular changes induced in the cat by coronary occlusion. Circulation Research, 39: 840-846.
- 25. Shell WE, Kjekshus JK & Sobel BE (1971). Quantitative assessment of the extent of myocardial infarction in the conscious dog by means of analysis of serial changes in serum creatine phosphokinase activity. Journal of Clinical Investigation, 50: 2614-2626.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
30 Jan 2004 -
Date of issue
Feb 2004
History
-
Accepted
14 Oct 2003 -
Received
25 Feb 2002