Abstract
8-Methoxy psoralen (8-MOP) exerts a short-term (24 h) mitogenic action, and a long-term (48-72 h) anti-proliferative and melanogenic action on two human melanoma cell lines, SK-Mel 28 and C32TG. An increase of intracellular calcium concentration was observed by spectrofluorometry immediately after the addition of 0.1 mM 8-MOP to both cell lines, previously incubated with calcium probe fluo-3 AM (5 µM). The intracellular Ca2+ chelator BAPTA/AM (1 µM) blocked both early (mitogenic) and late (anti-proliferative and melanogenic) 8-MOP effects on both cell lines, thus revealing the importance of the calcium signal in both short- and long-term 8-MOP-evoked responses. Long-term biological assays with 5 and 10 mM tetraethylammonium chloride (TEA, an inhibitor of Ca2+-dependent K+ channels) did not affect the responses to psoralen; however, in 24-h assays 10 mM TEA blocked the proliferative peak, indicating a modulation of Ca2+-dependent K+ channels by 8-MOP. No alteration of cAMP basal levels or forskolin-stimulated cAMP levels was promoted by 8-MOP in SK-Mel 28 cells, as determined by radioimmunoassay. However, in C32TG cells forskolin-stimulated cAMP levels were further increased in the presence of 8-MOP. In addition, assays with 1 µM protein kinase C and calcium/calmodulin-dependent kinase inhibitors, Ro 31-8220 and KN-93, respectively, excluded the participation of these kinases in the responses evoked by 8-MOP. Western blot with antibodies anti-phosphotyrosine indicated a 92% increase of the phosphorylated state of a 43-kDa band, suggesting that the phosphorylation of this protein is a component of the cascade that leads to the increase of tyrosinase activity.
8-[Methoxy]-psoralen; SK-Mel 28 cell line; C32TG cell line; Cell proliferation; Cell signaling; Melanoma
Braz J Med Biol Res, April 2004, Volume 37(4) 559-568
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells
M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci
Departamento de Fisiologia, Instituto de Biociências, Universidade de São Paulo, São Paulo, SP, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
8-Methoxy psoralen (8-MOP) exerts a short-term (24 h) mitogenic action, and a long-term (48-72 h) anti-proliferative and melanogenic action on two human melanoma cell lines, SK-Mel 28 and C32TG. An increase of intracellular calcium concentration was observed by spectrofluorometry immediately after the addition of 0.1 mM 8-MOP to both cell lines, previously incubated with calcium probe fluo-3 AM (5 µM). The intracellular Ca2+ chelator BAPTA/AM (1 µM) blocked both early (mitogenic) and late (anti-proliferative and melanogenic) 8-MOP effects on both cell lines, thus revealing the importance of the calcium signal in both short- and long-term 8-MOP-evoked responses. Long-term biological assays with 5 and 10 mM tetraethylammonium chloride (TEA, an inhibitor of Ca2+-dependent K+ channels) did not affect the responses to psoralen; however, in 24-h assays 10 mM TEA blocked the proliferative peak, indicating a modulation of Ca2+-dependent K+ channels by 8-MOP. No alteration of cAMP basal levels or forskolin-stimulated cAMP levels was promoted by 8-MOP in SK-Mel 28 cells, as determined by radioimmunoassay. However, in C32TG cells forskolin-stimulated cAMP levels were further increased in the presence of 8-MOP. In addition, assays with 1 µM protein kinase C and calcium/calmodulin-dependent kinase inhibitors, Ro 31-8220 and KN-93, respectively, excluded the participation of these kinases in the responses evoked by 8-MOP. Western blot with antibodies anti-phosphotyrosine indicated a 92% increase of the phosphorylated state of a 43-kDa band, suggesting that the phosphorylation of this protein is a component of the cascade that leads to the increase of tyrosinase activity.
Key words: 8-[Methoxy]-psoralen, SK-Mel 28 cell line, C32TG cell line, Cell proliferation, Cell signaling, Melanoma
Introduction
Psoralens are natural or synthetic planar furocoumarin which, in combination with ultraviolet light (UVA; 320-400 nm), have been used in the photochemical treatment of a variety of skin diseases including vitiligo, a hypopigmentary disorder, and psoriasis, a hyperproliferative disorder of the keratinocytes (1). The potent modulation of epidermal cell growth and differentiation promoted by psoralens in association with ultraviolet radiation (PUVA) has been attributed to the drug insertion in the DNA and the formation of photoadducts (2,3). It is now known that, in addition to photoadduct formation with DNA pyrimidine bases, PUVA also favors the binding of psoralens with other molecules such as membrane lipids, proteins and RNA (4).
Some investigators, however, have demonstrated that the action of psoralen can be exerted independent of radiation. In murine melanoma cells S-91 and B16, 8-methoxypsoralen (8-MOP) increases the activity of the melanogenic key enzyme, tyrosinase, in the absence of light (5). We have previously reported that 8-MOP elicits a biphasic response in two human melanoma cell lines, SK-Mel 28 and C32TG, which exhibited increased proliferation after the first 24 h treatment, followed by a decrease over the subsequent 48 h and thereafter. Cells treated with 8-MOP failed to enter the G1 phase, and did not exhibit any features of apoptosis. In addition, chronic exposure to 8-MOP in the absence of UVA enhanced tyrosinase activity in both cell lines (6).
The existence of low and high affinity sites for psoralens was demonstrated in a variety of cell lines (7,8) including the two human melanoma lines (6), and in the presence of UVA, 8-MOP covalently binds to sites other than DNA (6).
Despite the wide therapeutical use of psoralens, little is known about their mechanisms of action. Psoralens have been reported to block ATP-dependent potassium channels (9) and to intensify chloride secretion triggered by cholinomimetics after the opening of potassium channels by the increase of intracellular Ca2+ levels (10). Albrightson et al. (11) reported an increase of cAMP in normal human fibroblasts, epithelial cells and mononuclear leukocytes by 8-MOP. Mengeaud and Ortonne (12) demonstrated that 5-MOP promotes a small increase of cAMP concentration and, in addition, activates protein kinase C (PKC) in a murine melanoma line.
We report here the participation of calcium, calcium-activated K+-channels and a tyrosine-kinase in the intracellular signaling stimulated by 8-MOP in two human melanoma cell lines, SK-Mel 28 (melanotic) and C32TG (amelanotic).
Material and Methods
The human melanotic melanoma cell line, SK-Mel 28, was purchased from the American Type Culture Collection Repository (Rockville, MD, USA), and the human amelanotic melanoma line, C32TG, was provided by the Ludwig Institute for Cancer Research (São Paulo, SP, Brazil). Both cell lines were maintained as monolayer cultures in RPMI-1640 medium supplemented with 10% fetal calf serum (Cultilab, São Paulo, SP, Brazil), 2.0 g/l NaHCO3, 15 nM HEPES, and 1 mM sodium pyruvate, in the absence of antibiotics, at 37ºC and 5% CO2.
8-MOP, RPMI-1640, digitonin, forskolin, isobutylmethylxanthine (IBMX), tetraethylammonium chloride (TEA), sodium pyruvate, ethylenediaminetetraacetic acid (EDTA), Tris-HCl, probenecid, tapsigargin, dimethylsulfoxide, albumin, 2ß-mercaptoethanol, antipain, aprotinin, leupeptin, phenylmethanesulfonyl fluoride, bromophenol blue, phenol, glycerol, and activated charcoal were purchased from Sigma (St. Louis, MO, USA). 1,2-bis-(o-aminophenoxy)-ethane-N,N,N',N'-tetraacetic acid, tetraacetoxymethyl ester (BAPTA/AM), 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl) amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN-93) and 3-[1-[3-(amidinothio)propyl-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl) (Ro 31-8220) were acquired from Calbiochem (La Jolla, CA, USA). L-[3',5'-3H]-tyrosine (specific activity 57 Ci/mmol) and acrylamide were acquired from Amersham-Pharmacia (Buckinghamshire, England). X-ray film (Scientific Imaging Film X-OMAT AIR 13 x 18 cm2) was purchased from Kodak.
Determination of intracellular cAMP
Triplicates of 1.5 x 105 cells/well (in 6-well plates) were pre-incubated with RPMI medium containing 0.1 mM IBMX for 10 min at 37ºC. The cells were then incubated for 30 min at 37ºC, in 1 ml of RPMI medium containing 0.1 mM IBMX, plus one of the following treatments: control, 1 µM forskolin (an adenylyl cyclase activator), 1 µM forskolin + 0.1 mM 8-MOP, 0.1 mM 8-MOP, 10 µM forskolin, and 10 µM forskolin + 0.1 mM 8-MOP.
The reaction was stopped by the addition of cold ethanol. The medium was removed, the cells were scraped and the lysate transferred to Eppendorf tubes on ice. After 30 min, the samples were centrifuged (5,000 g, 10 min at 4oC) and the supernatant was transferred to test tubes and evaporated with nitrogen to a final volume of approximately 500 µl. Duplicates of each sample were used for determination of protein (bicinchoninic acid, BCA, protein assay kit, Pierce, Rockford, IL, USA) and cAMP concentrations (NEK-033; New England Nuclear Life Science Products, Boston, MA, USA). The cAMP radioimmunoassay is based on the competition between a radioactive antigen and the non-radioactive native antigen for a certain number of antibody binding sites. As a consequence, decreasing amounts of labeled antigen bind to the antibody as the amount of native cAMP increases. cAMP concentrations were calculated as pmol/mg protein and compared by Kruskal-Wallis non-parametric ANOVA.
Determination of intracellular calcium
SK-Mel 28 and C32TG melanoma cells were suspended (107/ml) in RPMI supplemented with 2% fetal bovine serum, and incubated with 5 µM fluo-3 AM (4-(6-acetoxymethoxy-2,7-dichloro-3-oxo-9-xanthenyl)-4'-methyl -2,2'(ethylenedioxy) dianiline-N,N,N',N'-tetraacetic acid tetrakis (acetoxymethyl) ester; Sigma) and 3 mM probenecid for 50 min at 37oC, with shaking. Aliquots (50 µl) were centrifuged (7,000 g), rinsed (3 x 500 µl) in Ca2+-free M buffer (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM D-glucose, and 50 mM HEPES, pH 7.2), and readings at 505 nm excitation and 530 nm emission were obtained with a spectrofluorometer (Hitachi, model F4500, Tokyo, Japan). The addition of 0.1 mM 8-MOP was followed by the addition of 12 µM tapsigargin, 10 µM digitonin, 12 µM EGTA, and 1 mM CaCl2 to determine the integrity of the cells.
Biological assays in the presence of inhibitors
SK-Mel 28 (1.5 x 105) or C32TG (3.0 x 105) cells were seeded in triplicate in 25-cm2 flasks containing RPMI-1640 medium supplemented as described above. After 24 h, the medium was replaced with fresh medium containing 0.1 mM 8-MOP or the solvent (0.1% ethanol). Each inhibitor (KN-93, Ro 31-8220, BAPTA/AM and TEA) was added 30 min before the addition of psoralen. The assays with kinase inhibitors lasted 72 h with medium changes every other day; an additional 24-h assay was introduced for TEA. Twenty-four hours before ending the experiment, 1 µCi/ml [3H]-tyrosine was added to each flask. At the end, tyrosinase activity was determined in the medium on the basis of the amount of 3H2O produced in the first step of melanogenesis by the method of Pomerantz (13), modified according to Mengeaud and Ortonne (12). Duplicate 200-µl medium samples from each flask were added to 800 µl of 10% activated charcoal in 10% trichloracetic acid. After vigorous shaking, the mixture was incubated at 9ºC for 20 min and centrifuged (7,000 g, 20 min), the supernatant (600 µl) was added to 3 ml of scintillation fluid and radioactivity was measured with a scintillation counter (Packard, model TRI-CARB 2100 TR, Downers Grove, IL, USA). The cells were harvested with Tyrode/EDTA solution and counted with the aid of a hemocytometer.
Tyrosinase activity (cpm/106 cells) and proliferation are reported as percentage of control (100%). The bioassay results were analyzed by ANOVA followed by the Student-Newman-Keuls test, and the experimental values were considered to be significantly different from control values at P £ 0.05.
Tyrosine-phosphorylation assay
After seeding (1 x 107 cells/80 cm2 flask) and adhesion, SK-Mel 28 cells were treated with 0.1 mM 8-MOP for 30 min. The medium was discarded and the cells were rinsed twice with sodium phosphate-buffered saline (PBS), harvested with Tyrode/EDTA solution, pelleted and resuspended, washed and centrifuged twice with 1 ml of cold PBS solution. After lysis, 10-µl aliquots were removed for protein determination with a BCA protein assay kit (Pierce-23225), and equivalent volumes containing 50 µg of protein were added to the same volume of sample buffer supplemented with 5% (v/v) 2ß-mercaptoethanol. After denaturation, the samples were run on a 5 to 20% acrylamide gel gradient (10 mA) and transferred to a 0.45-µM nitrocellulose membrane (15 V, 60 min). The membrane was then blocked for 2 h at 4ºC, in Tris-buffered saline with 0.1% Tween 2 (TBST) supplemented with 5% albumin solution and 0.01% sodium azide and incubated for 2 h at 4ºC with a mouse monoclonal primary antibody (IgG2a) P-Tyr (Py6a; 1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA) in TBST solution supplemented as above. After removal of the antibody, the membrane was rinsed (3 x 10 min) with TBST solution and incubated (30 min at 37ºC) with the anti-mouse IgG secondary antibody (1:1000, Calbiochem) in TBST containing 5% albumin. The membrane was then rinsed in TBST (3 x 10 min), the bands were detected by incubation (1 min) in luminol/enhancing detection solution (1:1, Amersham-Pharmacia), and the membrane photographed by contact (5 min) with X-ray film. The relative optical densities were determined by NIH Image software, version 1.62 (National Institute of Health, Bethesda, MD, USA).
Results
We have previously demonstrated that 8-MOP increased melanogenesis and decreased proliferation when incubated for a long period of time (72 h) in the absence of UV light. Interestingly enough, it exhibited a mitogenic effect after the first 24 h (6). We therefore decided to investigate the mechanisms of actions of 8-MOP in eliciting these responses.
Intracellular cAMP
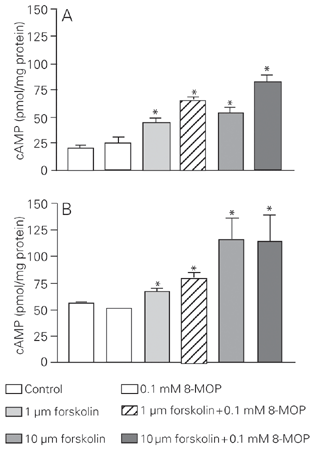
We first investigated the variation of cAMP levels in 8-MOP-treated and untreated cells on the basis of published data (11,12). As expected, 1 and 10 µM forskolin (positive controls) promoted an increase of cAMP concentration in both cell lines. The treatment with 0.1 mM 8-MOP did not cause any significant alteration in the basal concentration of cAMP or in the forskolin-stimulated levels in SK-Mel 28 cells (Figure 1A), thus excluding the role of a cAMP-activated protein kinase A in the 8-MOP-elicited responses. However, in C32TG cells, 8-MOP increased forskolin-stimulated levels, but not the basal concentration of cAMP (Figure 1B).
Effect of 8-MOP and forskolin on cAMP concentrations of SK-Mel 28 cells (A) and C32TG cells (B). The incubations were carried out for 30 min and when forskolin and 8-MOP were combined, the drugs were added simultaneously. Data are reported as means ± SEM for N = 6 experiments. *P £ 0.05 compared to control (Kruskal-Wallis nonparametric ANOVA test).
Role of intracellular calcium and Ca2+-activated K+ channels
An increase in intracellular calcium was observed in both cell lines immediately after the addition of 0.1 mM 8-MOP. The magnitude of the fluorescence increase was much higher in C32TG cells (Figure 2) than in SK-Mel 28 cells (Figure 3).
Our next step was to investigate the participation of Ca2+ in the long-term responses evoked by psoralen (decrease of proliferation and increase of tyrosinase activity). Table 1 shows that both responses exhibited by SK-Mel 28 and C32TG cells after 72 h of 8-MOP treatment were inhibited in the presence of 1 µM BAPTA/AM (an intracellular Ca2+ chelator). However, the effectiveness of BAPTA was greater in SK-Mel 28 cells than in C32TG cells, in which the responses were not totally reversed.
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells. M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (4): 559, 2004.
The increase of intracellular Ca2+ concentration has often been described in the literature as an initial signal for cell proliferation. Our previous results revealed that a 24-h treatment with 8-MOP stimulated the cells to proliferate (6). In an attempt to demonstrate the need for Ca2+ not only for this early proliferative peak, but also for the late decrease in proliferation and increase in tyrosinase activity, the cells were treated with BAPTA/AM only during the first 24 h of the assay (a total 8-MOP assay time of 72 h). The inhibition of the 8-MOP-evoked decrease of proliferation and increase of tyrosinase activity in SK-Mel 28 cells by BAPTA/AM (Table 2) indicates that an early rise in Ca2+ is required for both the early and late biological responses promoted by 8-MOP.
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells. M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (4): 559, 2004.
K+ channels have been reported to be responsible for the control of cell volume, a decisive function for the event of cell duplication. Having this in mind, we investigated the participation of Ca2+-dependent K+ channels in the biological responses evoked by 8-MOP.
TEA (5 and 10 mM) had no effect of its own, nor did it affect the responses of proliferation and tyrosinase activity in SK-Mel 28 cells after 72 h treatment with 0.1 mM 8-MOP (Table 3). In contrast, the proliferative effect observed after 24 h of treatment with 0.1 mM 8-MOP was reduced by 10 mM TEA (Table 4), suggesting that the short-term response of increased proliferation depends on the activity of Ca2+-modulated K+ channels.
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells. M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (4): 559, 2004.
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells. M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (4): 559, 2004.
Representative fluo-3 fluorescence tracing of C32TG cells. 1, 12 µM tapsigargin; 2, 0.1 mM 8-MOP; 3, 10 µM digitonin; 4, 12 µM EGTA. Cells were suspended in Ca2+-free M buffer and an 8-MOP stock solution was prepared with propylene glycol. Excitation = 505 nm; emission = 530 nm.
Representative fluo-3 fluorescence tracing of SK-Mel 28 cells. 1, 0.1 mM 8-MOP; 2, 12 µM tapsigargin; 3, 10 µM digitonin; 4, 1 mM Ca2+ as calcium chloride; 5, 12 µM EGTA. Cells were suspended in Ca2+-free M buffer and an 8-MOP stock solution was prepared with propylene glycol. Excitation = 505 nm; emission = 530 nm.
Biological activity of 8-MOP in the absence or presence of kinase inhibitors
In view of the important role of Ca2+ in the responses triggered by 8-MOP, we next investigated the participation of calcium-dependent kinases. PKC inhibition by Ro 31-8220 did not interfere with the biological activity of 0.1 mM 8-MOP in SK-Mel 28 cells. Furthermore, Ro 31-8220 caused a reduction in proliferation by itself, which was not accompanied by increased melanogenesis (Table 5).
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells. M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (4): 559, 2004.
KN-93, like Ro 31-8220, had no effect on the biological activity of 0.1 mM 8-MOP, and also caused a reduction in proliferation on its own, resulting in an enhancement of the 8-MOP-evoked response (Table 6). These data suggest that PKC and calcium-calmodulin-dependent kinase are necessary for the cell cycle to progress, but play no role in the responses promoted by 8-MOP.
The role of calcium, calcium-activated K+ channels, and tyrosine/kinase in psoralen-evoked responses in human melanoma cells. M.C. Isoldi, E.A. Pereira, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (4): 559, 2004.
Tyrosine-phosphorylation assays
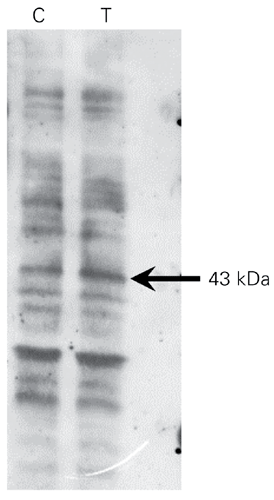
As cAMP-dependent protein kinase A (PKA) and calcium-dependent kinases do not seem to participate in the biochemical pathways activated by 8-MOP, we next investigated the participation of tyrosine kinases in the process. A 43-kDa protein shows a 92% increased phosphorylation on tyrosine after a 30-min treatment of SK-Mel 28 cells with 0.1 mM 8-MOP (Figure 4), thus demonstrating that a tyrosine kinase is activated upon contact of 8-MOP with the cells.
Western blot of tyrosine-phosphorylated proteins in SK-Mel 28 cells. The arrow indicates the 43-kDa phosphorylated protein (92% increase) after treatment with 0.1 mM 8-MOP for 30 min. C = controls; T = 0.1 mM 8-MOP.
Discussion
PKA and PKC have been mentioned by some investigators as being responsible for the effects of psoralen. Albrightson et al. (11) reported an 8-MOP-promoted increase of cAMP in normal human fibroblasts, epithelial cells, and mononuclear leukocytes in vitro. In addition, Mengeaud and Ortonne (12) demonstrated that 5-MOP induces a small increase of cAMP concentration and activates PKC in murine melanoma cells, since calphostin (a specific PKC inhibitor) reverses the increase of tyrosinase activity in response to 5-MOP. We have found no significant differences in cAMP concentration between untreated and psoralen-treated cells, suggesting that cAMP and cAMP-dependent PKA are probably not involved in the responses evoked by 8-MOP in human melanoma cells. The increase of forskolin-stimulated cAMP levels induced by 8-MOP in C32TG might well be due to the inhibition of a calcium-sensitive phosphodiesterase or stimulation of a calcium-sensitive adenylyl cyclase. In fact, in these cells the calcium increase evoked by 8-MOP was much higher than in SK-Mel 28 cells. And this might also be the reason for the lower inhibitory effectiveness of BAPTA in C32TG cells compared to SK-Mel 28.
Elevation of intracellular Ca2+ has frequently been reported as a signal for cell proliferation (14). Extracellular calcium is necessary to begin and to support mitosis, and the passage from G0 to G1 only occurs if there is an increase of intracellular Ca2+ (15). These last investigators (15) verified that mitogens promote bursts of increases in intracellular Ca2+ that, depending on cell type, may result in bursts of Ca2+/calmodulin production and/or PKC activation. The initial mitogen signal quickly appears (seconds to minutes) and is of short duration (minutes), but gives origin to long-lasting signals that can involve Ca2+-induced release of Ca2+ from the cell compartments (14). The elevation of intracellular Ca2+ levels by 8-MOP seems, at first, to contradict its anti-proliferative action. However, the growth curves revealed that a growth peak (@30%) appears 24 h after the introduction of 0.1 mM 8-MOP, followed by a significant reduction of proliferation (48 and 72 h) compared to the controls (6). Spectrofluorometric calcium profiles revealed that an immediate Ca2+ increase occurs with the addition of 8-MOP to the cell suspension. In addition, biological assays with 1 µM BAPTA/AM demonstrated the importance of Ca2+ participation in the responses promoted by 8-MOP in both cell lines. In these assays, 1 µM BAPTA/AM showed low cell toxicity and totally abolished the early and late biological responses evoked by psoralen. The initial Ca2+ signal seems to be also necessary for the second phase of the 8-MOP-evoked response, although it is an inhibition of proliferation. The presence of BAPTA/AM only in the first 24 h of the experiment was also enough to prevent the 8-MOP-evoked increase of tyrosinase activity, thus confirming that the initial burst of Ca2+ is essential for the second phase of 8-MOP activity.
We then considered the possibility that 8-MOP may modulate K+ channels of the human melanoma cells. Recent work has demonstrated the importance of potassium channels (voltage dependent, ATP sensitive and Ca2+ dependent) for the modulation of cell proliferation (16). Nilius and Wohlrab (17) reported that the proliferation of human melanoma IGR1 cells was inhibited by the blockade of Ca2+-dependent potassium channels. In SK-Mel 28 cells, the blockade of two types of K+ channels, Ca2+-activated and voltage rectifying, also resulted in proliferation inhibition (18). Nilius and Wohlrab (17) suggested that the hyperpolarization promoted by Ca2+-activated potassium channels would lead to an increase of Ca2+ influx in a positive feedback process. The hyperpolarization would also facilitate the rate of Na+ transport, thus modulating the cell volume and the reception of essential molecules for proliferation (16). In long-term biological assays (72 h) with 8-MOP in the presence of TEA (an inhibitor of Ca2+-dependent K+ channels), no alteration of the responses (proliferation inhibition and melanogenesis increase) to 8-MOP was observed, nor did TEA (5 and 10 mM) affect cell proliferation or melanogenesis on its own. In the short-term assay (24 h), however, the proliferation increase induced by 8-MOP was abolished in the presence of TEA. These results indicate that the Ca2+ signal evoked by 8-MOP induces the opening of Ca2+-dependent K+ channels, translated as an initial incentive to proliferation. In the late phase, Ca2+ would also be signaling proliferation inhibition, now independently of K+ channel activation.
The confirmation of Ca2+ participation in the responses promoted by 8-MOP directed our interest to the role of Ca2+-dependent kinases in the process. Biological assays in the presence of Ro 31-8220 (a PKC inhibitor) demonstrated that the inhibition of PKC in SK-Mel 28 cells promotes the inhibition of cell proliferation, without alteration of tyrosinase activity, and that the responses to 8-MOP were not diminished by the blocker. Indeed, the inhibition of proliferation was further increased in the presence of Ro 31-8220. Therefore, in this cell line, PKC activity seems to be related to the stimulation of proliferation, similarly to what was reported for normal melanocytes (19), without evoking an increase in melanogenesis. Similarly to Ro 31-8220, KN-93 (a calcium/calmodulin-dependent kinase inhibitor), exhibited an intrinsic anti-proliferative activity and did not affect the responses to 8-MOP.
Western blot of phosphorylated tyrosine revealed the increased phosphorylation of a 43-kDa protein after 0.1 mM 8-MOP treatment, demonstrating for the first time the participation of a tyrosine kinase in the mechanism of action of a psoralen. It is interesting to mention that the potent melanogenic hormone, a-MSH, promotes phosphorylation of a 43-kDa protein in murine melanoma S91 cells (20), thus suggesting that the phosphorylation of this protein is a component of the cascade that leads to the increase of tyrosinase activity. This 43-kDa protein could be a dimer of tyrosinase, and it is well known that tyrosinase phosphorylation at serine/threonine sites precedes its activation by a-MSH and other melanotropic agents (19). It is possible that phosphorylation of tyrosine sites in the enzyme by 8-MOP would also lead to increased activity. If the tyrosine-kinase activated by 8-MOP is a component of its own binding site, and whether it is intracellular or it is located in the membrane remains to be clarified. Since its activation also results in inhibition of proliferation, its nature probably differs from that of the tyrosine kinase receptors of growth factors.
Acknowledgments
We are thankful to Telma Pazini and Marcio Lima for technical assistance.
Address for correspondence: A.M.L. Castrucci, Departamento de Fisiologia, Instituto de Biociências, USP, 05508-900 São Paulo, SP, Brasil. Fax: +55-11-3091-7422. E-mail: amdlcast@usp.br
Research partially supported by CNPq (Nos. 522969/96-8 and 550676/2002-3) and FAPESP (Nos. 96/6409-0 and 01/02460-1). M.C. Isoldi and E.A. Pereira were research fellowships of FAPESP. Received May 8, 2003. Accepted January 5, 2004.
- 1. Laskin JD (1994). Cellular and molecular mechanisms in photochemical sensitization: studies on the mechanism of action of psoralens. Food and Chemical Toxicology, 32: 119-127.
- 2. Gasparro FP (1988). Immunological assay of 8-MOP photoadducts in DNA. Journal of Photochemistry and Photobiology, B22: 286-288.
- 3. Averbeck D (1989). Recent advances in psoralen phototoxicity mechanism. Photochemistry and Photobiology, 50: 859-882.
- 4. Zarebska Z (1994). Cell membrane, a target for PUVA therapy. Journal of Photochemistry and Photobiology, B23: 101-109.
- 5. Marwan MM, Jiang JW, Castrucci AML & Hadley ME (1990). Psoralens stimulate mouse melanocyte and melanoma tyrosinase activity in the absence of ultraviolet light. Pigment Cell Research, 3: 214-221.
- 6. Isoldi MC, Scarparo AC, Schumacher RI & Castrucci AML (1999). Psoralen activity and binding sites in melanotic and amelanotic human melanoma cells. Pigment Cell Research, 12: 367-375.
- 7. Laskin JD, Lee E, Yurkow EJ, Laskin DL & Gallo MA (1985). A possible mechanism of psoralen phototoxicity not involving direct interaction with DNA. Proceedings of the National Academy of Sciences, USA, 82: 6158-6162.
- 8. Yurkow EJ & Laskin JD (1987). Characterization of a photoalkylated psoralen receptor in HeLa cells. Journal of Biological Chemistry, 262: 8439-8442.
- 9. Szewczyk A, De Weille JR & Lazdunski M (1992). 8-Methoxypsoralen blocks ATP-sensitive potassium channels and stimulates insulin release. European Journal of Pharmacology, 216: 323-326.
- 10. Devor DC, Singh AK, Bridges RJ & Frizzell RA (1997). Psoralens: novel modulators of Cl- secretion. American Journal of Physiology, 272: C976-C988.
- 11. Albrightson CR, Fertel RH, Brown BV & Stephens R (1985). Psoralens increase the concentration of cyclic AMP in human cells in vitro Journal of Investigative Dermatology, 85: 264-268.
- 12. Mengeaud V & Ortonne JP (1994). Regulation of melanogenesis induced by 5-methoxypsoralen without ultraviolet light in murine melanoma cells. Pigment Cell Research, 7: 245-254.
- 13. Pomerantz SH (1966). The tyrosine hydroxylase activity of mammalian tyrosinase. Journal of Biological Chemistry, 241: 161-168.
- 14. Lovisolo D, Distasi C, Antoniotti S & Munaron L (1997). Mitogens and calcium channels. News in Physiological Sciences, 12: 279-285.
- 15. Dubois JM & Rouzaire-Dubois B (1993). Role of potassium channels in mitogenesis. Progress in Biophysics and Molecular Biology, 59: 1-21.
- 16. Wonderlin WF & Strobl JS (1996). Potassium channels, proliferation and G1 progression. Journal of Membrane Biology, 154: 91-107.
- 17. Nilius B & Wohlrab W (1992). Potassium channels and regulation of proliferation of human melanoma cells. Journal of Physiology, 445: 537-548.
- 18. Lepple-Wienhues A, Berweck S, Bohmig M, Leo CP, Meyling B, Garbe C & Wiederholt M (1996). K+ channels and the intracellular calcium signal in human melanoma cell proliferation. Journal of Membrane Biology, 151: 149-157.
- 19. Abdel-Malek Z, Swope VB, Suzuki I, Akcali C, Harriger MD, Boyce ST, Urabe K & Hearing VJ (1995). Mitogenic and melanogenic stimulation of normal human melanocytes by melanotropic peptides. Proceedings of the National Academy of Sciences, USA, 92: 1789-1793.
- 20. de Graan PN, Brussaard AB, Gamboni G, Girard J & Eberle AN (1987). a-MSH-induced changes in protein phosphorylation of Cloudman S91 mouse melanoma cells. Molecular and Cellular Endocrinology, 51: 87-93.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
22 Apr 2004 -
Date of issue
Apr 2004
History
-
Accepted
05 Jan 2004 -
Received
08 May 2003