Abstract
Patients expressing estradiol receptors in melanoma cells have been reported to have a better prognosis. We therefore decided to investigate the in vitro effects of ß-estradiol and tamoxifen on the growth and tyrosinase activity of SK-Mel 23 human melanoma cells. Twenty-four-hour treatment with 0.4 nM ß-estradiol inhibited cell proliferation in 30% (0.70 ± 0.03 x 10(5) cells) and increased tyrosinase activity in 50% (7130.5 ± 376.5 cpm/10(5) cells), as compared to untreated cells (1.0 ± 0.05 x 10(5) cells and 4769 ± 25.5 cpm/10(5) cells, respectively). Both responses were completely (100%) blocked by 1 µM tamoxifen. Higher concentrations (up to 1.6 nM) or longer treatments (up to 72 h) did not result in a larger effect of the hormone on proliferation or tyrosinase activity. Competition binding assays demonstrated the presence of binding sites to [2,4,6,7-³H]-ß-estradiol, and that the tritiated analogue was displaced by the unlabeled hormone (1 nM to 100 µM, Kd = 0.14 µM, maximal displacement of 93%) or by 10 µM tamoxifen (displacement of 60%). ß-estradiol also increased the phosphorylated state of two proteins of 16 and 46 kDa, after 4-h treatment, as determined by Western blot. The absorbance of each band was 1.9- and 4-fold the controls, respectively, as determined with Image-Pro Plus software. Shorter incubation periods with ß-estradiol did not enhance phosporylation; after 6-h treatment with the hormone, the two proteins returned to the control phosphorylation levels. The growth inhibition promoted by estradiol may explain the better prognosis of melanoma-bearing women as compared to men, and open new perspectives for drug therapy.
SK-Mel 23 cell line; Human melanoma; ß-estradiol; Tamoxifen; Cell proliferation; Tyrosinase activity
Braz J Med Biol Res, June 2004, Volume 37(6) 901-905 (Short Communication)
Biological activity and binding of estradiol to SK-Mel 23 human melanoma cells
M.S.M.V. Sarti, M.A. Visconti and A.M.L. Castrucci
Departamento de Fisiologia, Instituto de Biociências, Universidade de São Paulo, São Paulo, SP, Brasil
 Text
Text
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Patients expressing estradiol receptors in melanoma cells have been reported to have a better prognosis. We therefore decided to investigate the in vitro effects of ß-estradiol and tamoxifen on the growth and tyrosinase activity of SK-Mel 23 human melanoma cells. Twenty-four-hour treatment with 0.4 nM ß-estradiol inhibited cell proliferation in 30% (0.70 ± 0.03 x 105 cells) and increased tyrosinase activity in 50% (7130.5 ± 376.5 cpm/105 cells), as compared to untreated cells (1.0 ± 0.05 x 105 cells and 4769 ± 25.5 cpm/105 cells, respectively). Both responses were completely (100%) blocked by 1 µM tamoxifen. Higher concentrations (up to 1.6 nM) or longer treatments (up to 72 h) did not result in a larger effect of the hormone on proliferation or tyrosinase activity. Competition binding assays demonstrated the presence of binding sites to [2,4,6,7-3H]-ß-estradiol, and that the tritiated analogue was displaced by the unlabeled hormone (1 nM to 100 µM, Kd = 0.14 µM, maximal displacement of 93%) or by 10 µM tamoxifen (displacement of 60%). ß-estradiol also increased the phosphorylated state of two proteins of 16 and 46 kDa, after 4-h treatment, as determined by Western blot. The absorbance of each band was 1.9- and 4-fold the controls, respectively, as determined with Image-Pro Plus software. Shorter incubation periods with ß-estradiol did not enhance phosporylation; after 6-h treatment with the hormone, the two proteins returned to the control phosphorylation levels. The growth inhibition promoted by estradiol may explain the better prognosis of melanoma-bearing women as compared to men, and open new perspectives for drug therapy.
Key words: SK-Mel 23 cell line, Human melanoma, ß-estradiol, Tamoxifen, Cell proliferation, Tyrosinase activity

In addition to acting on reproductive functions, estrogens are known to affect growth and differentiation of a variety of cell types. It has been reported that estrogens and progestins may elicit hyperpigmentary responses by the stimulation of melanogenesis within skin melanocytes (1). In fact, an increase in the transcription of tyrosinase, TRP-1 and TRP-2 (enzymes of the melanin synthesis pathway), has been reported in normal human melanocytes by several investigators (see Ref. 2).
The presence of high-affinity receptors for estradiol has been reported for 24 primary human melanomas (3). Patients expressing estradiol receptors in melanoma cells seem to have a better prognosis, suggesting a putative inhibitory action of estradiol on the growth of this tumor (4). More recently, a second estrogen receptor, ERß, cloned from the rat prostate, has been shown to possess high affinity for estradiol and to bind to the same DNA elements as does the classic receptor, ERa (5). Both receptors may form heterodimers and modulate gene transcription via AP-1, although the effects depend on the ligand and the receptor type (6).
The modulatory actions of estradiol on melanoma cells and its mechanism of action are still poorly understood. We present here evidence that estradiol promotes the phosphorylation of two proteins of 16 and 46 kDa, respectively, followed by growth inhibition and melanogenesis activation in SK-Mel 23 human melanoma cells.
The human melanoma cell line SK-Mel 23, kindly provided by Dr. Anthony Albino (Memorial Sloan Kettering Cancer Center, New York, NY, USA), was maintained in MCDB 153 medium (Sigma, St. Louis, MO, USA) supplemented with 5% fetal calf serum (after steroid extraction in 2.5% activated charcoal), in the absence of antibiotics, at 37ºC.
Twenty-four hours after seeding (105 cells/well on a 6-well plate), the medium was replaced with fresh medium containing 0.4, 0.8 or 1.6 nM ß-estradiol, and 0.7 µCi/ml [3H]-tyrosine, in triplicate. In a second protocol, the medium was replaced with fresh medium containing 0.4 nM estradiol, 1 µM tamoxifen, or both, and 0.7 µCi/ml [3H]-tyrosine, in triplicate. A second control received 0.1% ethanol (tamoxifen solvent).
After 24 h, 48 h or 72 h, the cells were harvested and counted with a hemocytometer, and tyrosinase activity was measured in the medium (see Ref. 7, modified according to Ref. 8). Briefly, 200-µl duplicates of each medium were added to 800 µl of 10% activated charcoal in 10% trichloroacetic acid. The mixture was vortexed vigorously, incubated at 9ºC for 20 min and centrifuged at 3000 g for 10 min. The supernatant solution (500 µl) was added to 5 ml scintillation fluid and counted.
SK-Mel 23 cells exhibited a statistically significant 30% decrease in proliferation (mean ± SEM, 0.70 ± 0.03 x 105 cells) and a 50% increase in tyrosinase activity (7130.5 ± 376.5 cpm/105 cells; ANOVA, followed by the Student-Newman-Keuls tests) after 24-h treatment with 0.4 nM ß-estradiol, as compared to control (1.0 ± 0.05 x 105 cells and 4769 ± 25.5 cpm/105 cells, respectively). Higher concentrations up to 1.6 nM or longer incubation periods (up to 72 h) with estradiol did not result in a larger effect of the hormone on proliferation or tyrosinase activity (Table 1).
Biological activity and binding of estradiol to SK-Mel 23 human melanoma cells. M.S.M.V. Sarti, M.A. Visconti and A.M.L. Castrucci. Brazilian Journal of Medical and Biological Research, 37 (6): 901, 2004.
An increase in human melanoma growth in response to estradiol has been reported by some investigators (9), whereas others have observed no response (10). However, the cited authors have used incubation periods longer than 24 h (10) or pharmacological concentrations (10-5 M) of the hormone (9). The data in the literature are also controversial regarding melanogenesis. Jee and co-workers (11) described a decrease in tyrosinase activity in normal human melanocytes after 4 days of 1 nM estradiol treatment, whereas others reported an increase in tyrosinase, TRP-1 and TRP-2 (2) transcription.
Melanogenesis is known to be stimulated by cyclic AMP-dependent protein kinase (PKA) or diacylglycerol dependent-kinase (PKC) pathway. Interestingly, PKA-stimulating agents such as cholera toxin and methylxanthines act synergistically with estradiol in human non-pigmentary cells (12). In addition, 17ß-estradiol and calcitriol activate G-protein-coupled phospholipase Cgin osteoblasts, suggesting the presence of steroid membrane receptors (13). This may be a pathway for the ß-estradiol-induced increase of tyrosinase activity in SK-Mel 23 cells.
However, both the anti-proliferative and melanogenic responses evoked by 0.4 nM estradiol were abolished in the presence of 1 µM tamoxifen, suggesting that estradiol probably exerted its effects through the classical estradiol receptor.
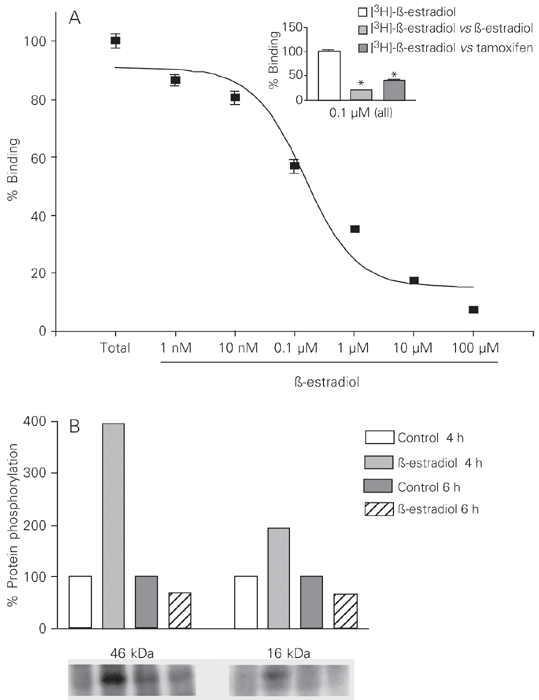
In view of these observations, we performed competition binding assays with [2,4,6,7-3H]-ß-estradiol. SK-Mel 23 cells were plated (2 x 105 cells/well on a 12-well plate) on MCDB medium supplemented with 5% charcoal-activated serum, and incubated in 0.1 µM [2,4,6,7-3H]-ß-estradiol and increasing concentrations (1 nM to 100 µM) of ß-estradiol or 10 µM tamoxifen for 45 min at 37ºC. The medium was removed, the cells were rinsed with 0.2% albumin-containing PBS, and lysed with 0.2% albumin and 0.5% Triton X-100 in PBS. Duplicates of 200 µl of the lysates were added to 5 ml of scintillation fluid and counted with a scintillation counter (model TriCarb 2100 TR Packard, Downers Grove, IL, USA). Analysis of the competition curve (Figure 1A) with the Graphpad Prism software indicated a single binding site with Kd = 0.14 µM. Maximal displacements of 93 and 60% were seen in the presence of 100 µM estradiol and 10 µM tamoxifen, respectively (Figure 1A).
Miller et al. (14), using immunocytochemistry, identified classic estrogen receptors in only 2 of 69 human melanomas. On the other hand, in addition to the classic receptor (Kd = 0.1 to 1.0 nM) (see, e.g., Ref. 15), another binding site, the so-called type II estrogen receptor, has been characterized in a variety of human melanomas (16). This site exhibits lower affinity for the ligand, but the same specificity, and also recognizes tamoxifen. Indeed, the significant anti-proliferative activity of ß-estradiol and selective analogues in normal and malignant tissues has been attributed to type II binding sites (17). Our data suggest that SK-Mel 23 human melanoma cells possess type II estradiol binding sites, since they exhibit low affinity for the ligand, and the hormone exerts an anti-proliferative action.
For the phosphorylation assays, 24 h after seeding (1 x 106 cells/80 cm2 flask), SK-Mel 23 cells were treated for 16 h with serum- and P-free RPMI 1640 (Gibco BRL, Rockville, MD, USA). Cells (5 x 105) were then resuspended in 100 µl containing 250 µCi/ml 32P (Amersham-Pharmacia, Buckinghamshire, England) at 37ºC for 2 h under constant shaking. Aliquots were incubated in 0.4 nM estradiol for 15, 30, and 60 min, and for 2, 4, and 6 h. The cells were then rinsed in cold PBS on ice (2X, 1 ml), pelleted and lysed in 50 µl RIPA buffer (1.2% aprotinin, 4 µM antipain, 5 mM EDTA, 5 µM leupeptin, 1.1 mM Na3VO4, 150 mM NaCl, 1.4 mM PMSF, 0.1% SDS, 10 mM Tris-HCl, 1% Triton X-100, pH 7.4; Sigma). The samples were vortexed vigorously, kept on ice for 15 min, and centrifuged at 2000 g for 10 min at 4ºC. Cold acetone was then added to the supernatant (5:1, v/v), which was kept at -20ºC overnight to precipitate the proteins.
The samples were then centrifuged at 2000 g for 10 min at 4ºC and the proteins resuspended in 50 µl buffer (5% v/v 2ß-mercaptoethanol, 0.002% bromophenol blue, 50% glycerol, 2% SDS, and 125 mM Tris-HCl, pH 6.8; Sigma). After heat denaturation, 1 µl of each sample was removed for radioactivity reading, and equal amounts of radioactivity were applied to a 5 to 20% gradient acrylamide gel which was electrophoresed at 10 mA for 1 h. The gel was then wrapped in cellophane and evaporated in hot air for 2 h for later contact (1-3 h) with X-ray film (Kodak scientific imaging film X-OMAT AIR 13 x 18 cm2). The relative densitometries were determined with the Image-Pro Plus software, version 4.5.1 (Media Cybernetics, Silver Spring, MD, USA).
The phosphorylation assays revealed that 0.4 nM estradiol increased the phosphorylated state of 2 proteins, of 16 and 46 kDa, respectively, after 4 h of treatment. The absorbance of each band was 1.9- and 4-fold the control density, respectively. Shorter incubation period (2 h) did not promote phosphorylation, and after 6 h of ß-estradiol treatment the 16- and 46-kDa substrates had been dephosphorylated (Figure 1B).
Reversible tyrosine phosphorylation of the estradiol receptor occurs after receptor activation by the ligand. The basal phosphorylation level of the receptor increases 3-4 times upon stimulation with estradiol or tamoxifen (18). However, the receptor is about 66 kDa, very different in size from the 16- and 46-kDa phosphorylated proteins in SK-Mel 23 cells.
It is well known that the melanizing hormone, a-MSH, promotes phosphorylation of tyrosinase, a 43-kDa protein, in murine and human melanocytes, by either PKA or PKC (19,20). The phosphorylation of the 46-kDa protein may well be a component of the cascade leading to the increase in tyrosinase activity promoted by ß-estradiol in SK-Mel 23 cells.
In conclusion, our data suggest the presence of type II estrogen binding sites in SK-Mel 23 cells which, once activated by estradiol, induce phosphorylation of 16- and 46-kDa proteins. These events lead to differentiation of melanoma cells, as there is a decrease of proliferation and an increase in melanogenesis.
A, Representative competition binding assay of 0.1 µM [2,4,6,7-3H]-ß-estradiol in the presence of increasing concentrations (1 nM to 100 µM) of ß-estradiol (N = 9), or in the presence of 10 µM ß-estradiol or 10 µM tamoxifen (insert, N = 3) in SK-Mel 23 melanoma cells. Data are reported as means ± SEM. B. Representative time-course phosphorylation assay of untreated and 0.4 nM ß-estradiol-treated SK-Mel 23 melanoma cells. Bar graph shows the values determined by densitometry which are compared to controls.
Address for correspondence: A.M.L. Castrucci, Departamento de Fisiologia, IB, USP, R. do Matão, Trav. 14, 05508-900 São Paulo, SP, Brasil. Fax: +55-11-3091-7422. E-mail: amdlcast@usp.br
Research partially supported by CNPq (Nos. 522969/96-8 and 550676/2002-3) and FAPESP (Nos. 96/6409-0 and 01/02460-1). M.S.M.V. Sarti was the recipient of a FAPESP fellowship. Received May 7, 2003. Accepted February 26, 2004.
- 1. Grimes PE (1995). Melasma: etiologic and therapeutic considerations. Archives of Dermatology, 131: 1453-1457.
- 2. Kippenberger S, Loitsch S, Solano F, Bernard A & Kaufmann R (1998). Quantification of tyrosinase, TRP-1, and TRP-2 transcripts in human melanocytes by reverse transcriptase-competitive multiplex PCR - regulation by steroid hormones. Journal of Investigative Dermatology, 110: 364-367.
- 3. Stoica A, Hoffman M, Marta L & Voiculetz N (1991). Estradiol and progesterone receptors in human cutaneous melanoma. Neoplasma, 38: 137-146.
- 4. Chaudhuri PK, Walker MJ, Beattie CW, Dasgupta TK & Patel MK (1985). Prognostic influence of oestrogen receptor in human melanoma. In: Bagnara J, Klaus SN, Paul E & Schartl M (Editors), Pigment Cell: Biological, Molecular and Clinical Aspects of Pigmentation University of Tokyo Press, Tokyo, Japan, 595-599.
- 5. Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S & Gustafsson J (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors a and ß. Endocrinology, 138: 863-870.
- 6. Paech K, Webb P, Kuiper GGJM, Nilsson S, Gustafsson JA, Kushner PJ & Scanlan TS (1997). Differential ligand activation of estrogen receptor ERa and ERß at AP1. Science, 277: 1508-1510.
- 7. Pomerantz SH (1969). L-tyrosine-3,5-3H assay for tyrosinase development in skin of newborn hamsters. Science, 164: 838-839.
- 8. Mengeaud V & Ortonne JP (1994). Regulation of melanogenesis induced by 5-methoxypsoralen without ultraviolet light in murine melanoma cells. Pigment Cell Research, 7: 245-254.
- 9. McClay EF, Jones JA, Winsk PJ, Albright KD, Christen RD & Howell SB (1996). Determinants of tamoxifen sensitivity control the nature of the synergistic interaction between tamoxifen and cisplatin. Cancer Research, 36: 3993-3997.
- 10. Lama G, Angelucci C, Bruzzese N, Iacopino F, Nori SL, D'Atris S, Turrizziani M, Bonmassar E & Sica G (1999). Sensitivity of human melanoma cells to oestrogens, tamoxifen and quercetin: is there any relationship with type I and type II oestrogen binding site expression? Melanoma Research, 8: 313-322.
- 11. Jee S, Lee S, Chiu H, Chang C & Chen TJ (1994). Effects of estrogen and estrogen receptor in normal human melanocytes. Biochemical and Biophysical Research Communications, 199: 1407-1412.
- 12. Cho H & Katzenellenbogen BS (1993). Synergistic activation of estrogen receptor-mediated transcription by estradiol and protein-kinase activators. Molecular Endocrinology, 7: 441-452.
- 13. Le Mellay V, Grosse B & Lieberherr M (1997). Phospholipase Cß and membrane action of calcitriol and estradiol. Journal of Biological Chemistry, 227: 11902-11907.
- 14. Miller JG, Gee J, Price A, Garbe C, Wagner M & MacNeil S (1998). Investigation of oestrogen receptors, sex steroids and soluble adhesion molecules in progression of malignant melanoma. Melanoma Research, 7: 187-208.
- 15. Viljoen TC, van Aswegen CH & du Plessis DJ (1995). Binding of estradiol to whole prostatic DU-145 cells in the presence and absence of tamoxifen and acetylsalicylic acid. Prostate, 27: 160-165.
- 16. Piantelli M, Maggiano N, Ricci R, Larocca LM, Capelli A, Scambia G, Isola G, Natali PG & Ranelletti OF (1995). Tamoxifen and quercetin interact with type II estrogen binding sites and inhibit the growth of human melanoma cells. Journal of Investigative Dermatology, 105: 248-253.
- 17. Markaverich BM & Alejandro MA (1998). Type II [3H]-estradiol binding site antagonists: inhibition of normal and malignant prostate cell growth and proliferation. International Journal of Oncology, 12: 1127-1135.
- 18. Le Goff P, Montano MM, Schodini DJ & Katzenellenbogen BS (1994). Phosphorylation of the human estrogen receptor. Journal of Biological Chemistry, 269: 4458-4466.
- 19. Abdel-Malek ZA, Swope VB, Suzuki I, Akcali C, Harriger MD, Boyce ST, Urabe K & Hearing VJ (1995). Mitogenic and melanogenic stimulation of normal human melanocytes by melanotropic peptides. Proceedings of the National Academy of Sciences, USA, 92: 1789-1793.
- 20. deGraan PN, Brussaard AB, Gamboni G, Girard J & Eberle AN (1987). a-MSH induces changes in protein phosphorylation of Cloudman S-91 mouse melanoma cells. Molecular and Cellular Endocrinology, 51: 87-93.
Publication Dates
-
Publication in this collection
28 May 2004 -
Date of issue
June 2004
History
-
Accepted
26 Feb 2004 -
Received
07 May 2003