Abstract
Oxidative stress and hepatic mitochondria play a role in the pathogenesis of nonalcoholic fatty liver disease. The aim of the present study was to evaluate the role of hepatic mitochondrial dysfunction and oxidative stress in the pathogenesis of the disease. Fatty liver was induced in Wistar rats with a choline-deficient diet (CD; N = 7) or a high-fat diet enriched with PUFAs-omega-3 (H; N = 7) for 4 weeks. The control group (N = 7) was fed a standard diet. Liver mitochondrial oxidation and phosphorylation were measured polarographically and oxidative stress was estimated on the basis of malondialdehyde and glutathione concentrations. Moderate macrovacuolar liver steatosis was observed in the CD group and mild liver steatosis was observed in the periportal area in the H group. There was an increase in the oxygen consumption rate by liver mitochondria in respiratory state 4 (S4) and a decrease in respiratory control rate (RCR) in the CD group (S4: 32.70 ± 3.35; RCR: 2.55 ± 0.15 ng atoms of O2 min-1 mg protein-1) when compared to the H and control groups (S4: 23.09 ± 1.53, 17.04 ± 2.03, RCR: 3.15 ± 0.15, 3.68 ± 0.15 ng atoms of O2 min-1 mg protein-1, respectively), P < 0.05. Hepatic lipoperoxide concentrations were significantly increased and the concentration of reduced glutathione was significantly reduced in the CD group. A choline-deficient diet causes moderate steatosis with disruption of liver mitochondrial function and increased oxidative stress. These data suggest that lipid peroxidation products can impair the flow of electrons along the respiratory chain, causing overreduction of respiratory chain components and enhanced mitochondrial reactive oxygen species. These findings are important in the pathogenesis of nonalcoholic fatty liver disease.
Hepatic mitochondrial dysfunction; Oxidative stress; Nonalcoholic fatty liver disease; Choline-deficient diet; High-fat diet
Braz J Med Biol Res, February 2006, Volume 39(2) 189-194
Liver mitochondrial dysfunction and oxidative stress in the pathogenesis of experimental nonalcoholic fatty liver disease
 Correspondence and Footnotes
Correspondence and Footnotes
 C.P.M.S. Oliveira1, A.M.M. Coelho2, H.V. Barbeiro3, V.M.R. Lima1, F. Soriano3, C. Ribeiro4, N.A.T. Molan2, V.A.F. Alves4, H.P. Souza3, M.C.C. Machado2 and F.J. Carrilho1
C.P.M.S. Oliveira1, A.M.M. Coelho2, H.V. Barbeiro3, V.M.R. Lima1, F. Soriano3, C. Ribeiro4, N.A.T. Molan2, V.A.F. Alves4, H.P. Souza3, M.C.C. Machado2 and F.J. Carrilho1
1Departamento de Gastroenterologia (LIM 07), 2Departamento de Cirurgia (LIM 37), 3Departamento de Emergência (LIM 51), 4Departamento de Patologia (LIM 14), Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Oxidative stress and hepatic mitochondria play a role in the pathogenesis of nonalcoholic fatty liver disease. The aim of the present study was to evaluate the role of hepatic mitochondrial dysfunction and oxidative stress in the pathogenesis of the disease. Fatty liver was induced in Wistar rats with a choline-deficient diet (CD; N = 7) or a high-fat diet enriched with PUFAs-w-3 (H; N = 7) for 4 weeks. The control group (N = 7) was fed a standard diet. Liver mitochondrial oxidation and phosphorylation were measured polarographically and oxidative stress was estimated on the basis of malondialdehyde and glutathione concentrations. Moderate macrovacuolar liver steatosis was observed in the CD group and mild liver steatosis was observed in the periportal area in the H group. There was an increase in the oxygen consumption rate by liver mitochondria in respiratory state 4 (S4) and a decrease in respiratory control rate (RCR) in the CD group (S4: 32.70 ± 3.35; RCR: 2.55 ± 0.15 ng atoms of O2 min-1 mg protein-1) when compared to the H and control groups (S4: 23.09 ± 1.53, 17.04 ± 2.03, RCR: 3.15 ± 0.15, 3.68 ± 0.15 ng atoms of O2 min-1 mg protein-1, respectively), P < 0.05. Hepatic lipoperoxide concentrations were significantly increased and the concentration of reduced glutathione was significantly reduced in the CD group. A choline-deficient diet causes moderate steatosis with disruption of liver mitochondrial function and increased oxidative stress. These data suggest that lipid peroxidation products can impair the flow of electrons along the respiratory chain, causing overreduction of respiratory chain components and enhanced mitochondrial reactive oxygen species. These findings are important in the pathogenesis of nonalcoholic fatty liver disease.
Key words: Hepatic mitochondrial dysfunction, Oxidative stress, Nonalcoholic fatty liver disease, Choline-deficient diet, High-fat diet
Introduction
Nonalcoholic fatty liver disease (NAFLD) involves fatty liver (hepatic steatosis) and nonalcoholic steatohepatitis (NASH) that progresses from hepatic steatosis with lobular inflammation to ballooning degeneration, fibrosis, and eventually to cirrhosis (1,2).
NAFLD is associated with several predisposing factors such as obesity, diabetes, dyslipidemia, jejunoileal bypass, drugs, and parenteral nutrition. However, the progression to fibrosis and cirrhosis is not known. Some studies have shown that liver injury is mediated by oxidative stress (3,4), endotoxins, cytokines (5,6), and hyperinsulinemia (7,8). Oxidative stress plays a central role in the pathogenesis of NASH. The increased production of reactive oxygen species (ROS) is known to cause lipid peroxidation, followed by an inflammatory response, and activation of stellate cells leading to fibrogenesis (9-11). Mitochondria are responsible for oxidative phosphorylation and fatty acid ß-oxidation and are the main source of cellular ROS. Therefore, dysfunction of liver mitochondria may play an important role in the induction of hepatic steatosis and NASH. The aim of the present study was to evaluate the role of liver mitochondrial dysfunction and oxidative stress in the pathogenesis of experimental fatty liver induced by different diets in an animal model.
Material and Methods
The study was designed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised in 1996) and the Guidelines of Animal Experimentation of the University of São Paulo School of Medicine, São Paulo, SP, Brazil, for the care and use of laboratory animals. Male Wistar rats weighing 300 to 350 g were housed in cages with a controlled 12-h light/dark cycle, receiving water ad libitum. Fatty liver was induced in these animals by two different diets: choline-deficient diet (CD; N = 7) or high-fat diet enriched with polyunsaturated fatty acids (PUFAs-w-3) (H; N = 7) for 4 weeks (Table 1). The control group (N = 7) was fed a standard diet. After 4 weeks, the rats were sacrificed under anesthesia with 0.2 mg/kg ketamine hydrochloride injected intraperitoneally and serum, plasma and liver samples were collected for biochemical analysis, histological examination, oxidative stress analysis, and analysis of mitochondrial function.
Biochemical analysis
Serum alanine aminotransferase (AST), aspartate aminotransferase (ALT), cholesterol, and triglycerides were analyzed by standard methods.
Histological analysis
Fragments of liver tissue previously fixed in 10% formalin solution were processed and stained with hematoxylin-eosin and Masson trichrome. The following histological variables were assessed and scored from 0 to 3 by a blinded experienced pathologist: macro- and microvacuolar fatty change, fatty zonal distribution, foci of necrosis, portal and perivenular fibrosis, as well as the inflammatory infiltrate and its zonal distribution.
Oxidative stress analysis
The thiobarbituric acid method was used to quantify lipid peroxidation in tissues, measured as thiobarbituric acid-reactive substances (TBARS). Tissues (100 mg/mL) were homogenized in 1.15% KCl buffer, and centrifuged at 14,000 g for 20 min. The supernatant was then stored at -70ºC. An aliquot of the supernatant was added to a reaction mixture of 1.5mL 0.8% thiobarbituric acid, 200 µL 8.1% (v/v) SDS, 1.5 mL 20% (v/v) acetic acid, pH 3.5, and 600 µL distilled H2O, and heated to 90ºC for 45 min. After cooling to room temperature, the samples were cleared by centrifugation at 10,000 g for 10min, and their absorbance was measured at 532 nm using 1,1,3,3-tetramethoxypropane as an external standard. The quantity of lipid peroxides is reported as nmol malondialdehyde (MDA) equivalents/mg protein. Glutathione (GSH) assay: tissues were homogenized (100 mg/mL) in 5% (v/v) sulfosalicylic acid. The homogenates were centrifuged at 10,000 g for 20 min, and an aliquot of the clear supernatant (20 mL) was combined with 0.3 M Na2HPO4 (160 mL) and 0.04% 5,59-dithiobis-(2-nitrobenzoic acid) in 1% sodium citrate (20 mL). After 10-min incubation at room temperature, absorbance was read at 405 nm in a Tecan microplate reader (Grödig, Salzburg, Austria). GSH concentrations were calculated from a standard curve constructed with known concentrations of reduced GSH and are reported as µg GSH/mg protein.
Oxidation and phosphorylation of liver mitochondria
Liver mitochondria were prepared as previously described (12). Mitochondrial oxygen consumption was measured polarographically (12) using a Gilson 5/6H Oxygraph (Gilson Medical Electronics, Inc., Middleton, WI, USA) in a closed reaction vessel fitted with a Clark oxygen electrode (Yellow Springs Instruments Co., Yellow Springs, OH, USA) at 28ºC. The respiratory control rate, considered to be an index of oxidative and phosphorylative mitochondrial function, was calculated as the rate of oxygen consumption in the presence of ADP (state 3, S3) over the consumption in the absence of ADP (state 4, S4). Respiratory states S3 and S4 were measured and reported as ng atoms of O2 min-1 mg protein-1. Mitochondria protein content was determined by the method of Lowry et al.
Statistical analysis
Data are reported as means ± SEM. Groups were compared by one-way analysis of variance (ANOVA), with the level of significance set at P < 0.05.
Results
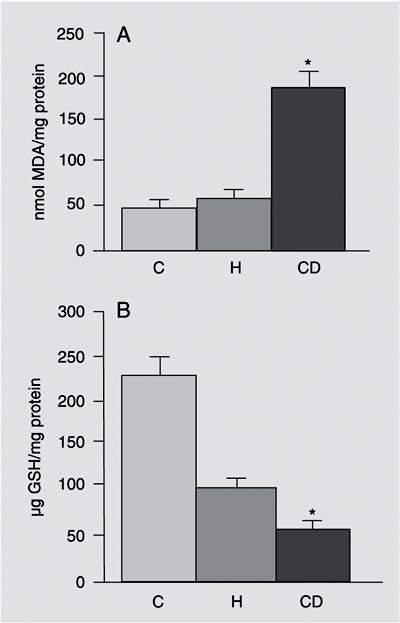
The CD diet caused moderate micro- and macrovacuolar liver steatosis in the periportal zone, while the H diet caused mild liver steatosis in the periportal zone. In both test groups, necroinflammatory activity was minimal. No fibrosis was seen in any of the groups. There was no significant difference in final body weight among groups and the diet composition was similar for all groups in terms of nutrients, except for fat and sucrose (Figure 1). AST and ALT levels were similar in the animals receiving both experimental diets compared to control. Cholesterol levels were decreased and triglyceride (TG) levels were increased in the CD group compared to control and H animals. CD animals presented an increase in oxygen consumption rate by mitochondria in S4, and a decrease in both respiratory control rate and ADP/oxygen ratio when compared to the H and control groups (Table 2). Basal respiration (S4), succinate dependent, was significantly higher in the CD group, resulting in a 92% average increase of oxygen consumption compared to control. Oxidative stress was increased in animals fed the CD diet: hepatic lipoperoxide (TBARS) concentrations were significantly increased and the concentration of reduced GSH was significantly lower in the CD group compared to the H and control groups (Figure 2A,B).
Initial and final weight of the animals. Data are reported as means ± SEM for 7 rats in each group as a function of diets. CD = choline-deficient diet; H = high-fat diet enriched with PUFAs-w-3; C = standard diet (control).
A, Thiobarbituric acid-reactive substances-malondialdehyde (MDA) equivalents in liver samples. Lipid peroxides are reported as nmol MDA/mg protein. B, Glutathione (GSH) assay in liver samples. GSH concentrations are reported as µg GSH/mg protein. Data are reported as means ± SEM for 7 rats in each group. CD = choline-deficient diet; H = high-fat diet; C = standard diet (control). *P < 0.05 for CD vs H and C (ANOVA).
Oxidation and phosphorylation of liver mitochondria in experimental nonalcoholic fatty liver disease.
Discussion
NAFLD is ascribed to an imbalance between the excessive uptake of free fatty acids by the liver with subsequent increase in TG synthesis, and the reduction of fatty acid oxidation and TG secretion (very low density lipoproteins, VLDLs) in the liver. Choline is an essential nutrient for the secretion of TGs (VLDLs) by the liver, and its privation impairs their transport, leading to fatty liver in the rat. A choline-deficient diet is a classic general model of NAFLD (13). On the other hand, lipogenesis is stimulated by a high carbohydrate diet, whereas it is inhibited by PUFAs and by fasting. The supply of PUFAs reduces the production and excretion of VLDL and decreases serum TG concentration (14). Sekiya et al. (15) demonstrated that animal PUFAs (fish oil) ameliorate hepatic steatosis through the suppression of sterol regulatory element-binding protein-1 in ob/ob mice. In the present study, the group that received a high-fat diet enriched with animal PUFA (fish oil) developed only a mild liver steatosis in the periportal zone and had normal plasma levels of cholesterol and TGs. These data confirm reports (14-16) that a high-fat diet enriched with PUFAs-w-3 (fish oil) has a protective effect on the liver, causing only mild liver steatosis. In animals fed the choline-deficient diet, liver steatosis was more severe and TG levels were also increased. These differences suggest that a high carbohydrate diet with an increase of TGs or impairment of VLDL exportation could play an important role in the pathogenesis of NAFLD.
Mitochondria are involved in fatty acid ß-oxidation and in oxidative phosphorylation and are an important cellular source of ROS. Therefore, mitochondrial dysfunction may play a central role in the accumulation of fat in the liver ("first hit") and the excessive production of ROS by mitochondria may result in lipid peroxidation ("second hit") (6). Drug-induced fatty liver has increased lipid peroxidation (17) and ob/ob mice have an increased ROS production (11). Our group has demonstrated in previous studies that the presence of steatosis correlates with an increase in superoxide anion hydroperoxide generation in animals fed a choline-deficient diet (18) and the use of vitamin C reduces liver steatosis and peroxidation (19). In the present study, lipid peroxidation was also increased and GSH was decreased in the CD group while the oxidative stress of animals fed a high-fat diet enriched with PUFAs-w-3 was similar to that observed in controls. The lack of lipid peroxidation in the H group could be explained by the fact that no steatosis developed. Additionally, chronic impairment of mitochondrial ß-oxidation could cause microvacuolar and/or macrovacuolar steatosis and an increase in ROS. Our results showed mitochondrial dysfunction with a marked succinate-dependent increase in basal respiration (S4) in CD animals, resulting in an average 92% increase of oxygen consumption when compared to the control group. This increased mitochondrial ROS formation may further oxidize fat deposits, causing a vicious cycle with more lipid peroxidation, more mitochondrial damage, and more ROS formation. ROS may oxidize fat deposits, releasing lipid peroxidation products that damage mitochondrial DNA and proteins to partially block the flow of electrons along the respiratory chain, thus further increasing mitochondrial ROS formation. ROS may also deplete antioxidants and cause the formation of tumor necrosis factor-w, two effects that may further impair the flow of electrons and increase mitochondrial ROS formation (20).
PUFAs-w-3 (fish oil) can be used by cyclooxygenase to produce prostaglandins of lower potency, that will induce less inflammation, or may reduce inflammation in some other way. Since PUFA-w-3 prostaglandins are less potent they induce less ROS production, a fact that may explain our findings.
There was no difference in mitochondrial function between the H diet and control. The high-fat diet caused only mild liver steatosis but mitochondrial function was similar to that observed in controls. According to this hypothesis, the increase in ROS production ("second hit") may result from the damage of mitochondrial ß-oxidation ("first hit"), leading to oxidative stress. Our study suggests that the pathogenesis of fatty liver differed between the two diets: the CD diet led to more severe, predominantly macrovacuolar steatosis, increased mitochondrial dysfunction, increased oxidative stress, and higher plasma TG levels than the H diet. The choline-deficient rat model is important for assessing oxidative stress independent of obesity, whereas the high fat diet model more closely resembles a leptin-resistant model. On the other hand, a high-fat diet enriched with polyunsaturated fat (w-3) slightly increases the arrival of fatty acids into the liver and, although producing mild steatosis, it preserves mitochondrial ß-oxidation and phosphorylation and the plasma triglyceride levels are similar to those in the control group.
The results of the present study suggest that the excessive arrival of fatty acid to the liver due to a high-fat diet enriched with polyunsaturated fat (w-3) may cause fatty liver but without mitochondrial dysfunction. Other mechanisms, such as a metabolic syndrome, may be more important than diet in the pathogenesis of NAFLD.
Publication supported by Programa de Apoio à Pós-Graduação-CAPES (PROAP). Received May 4, 2005. Accepted September 22, 2005.
- 1. Matteoni CA, Younossi ZM, Gramlich T et al. (1999). Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology, 116: 1413-1419.
- 2. Falck-Ytter Y, Younossi ZM, Marchesini G et al. (2001). Clinical features and natural history of nonalcoholic steatosis syndromes. Seminars in Liver Disease, 21: 17-26.
- 3. Chitturi S & Farrell GC (2001). Ethiopathogenesis of nonalcoholic steatohepatitis. Seminars in Liver Disease, 21: 27-41.
- 4. Mehta K, Van Thiel DH, Shah N et al. (2002). Nonalcoholic fatty liver disease: pathogenesis and the role of antioxidants. Nutrition Reviews, 60: 289-293.
- 5. Tilg H & Diehl AM (2000). Cytokines in alcoholic and nonalcoholic steatohepatitis. New England Journal of Medicine, 343: 1467-1476.
- 6. Wigg AJ, Roberts-Thomson IC, Dymock RB et al. (2001). The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut, 48: 206-211.
- 7. Luyckx FH, Lefebvre PJ & Scheen AJ (2000). Non-alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. Diabetes Metabolism, 26: 98-106.
- 8. Scheen AJ & Luyckx FH (2003). Nonalcoholic steatohepatitis and insulin resistance: interface between gastroenterologists and endocrinologists. Acta Clinica Belgica, 58: 81-91.
- 9. Yang S, Zhu H, Li Y et al. (2000). Mitochondrial adaptations to obesity-related oxidant stress. Archives of Biochemistry and Biophysics, 378: 259-268.
- 10. Curzio M, Esterbauer H & Dianzani MU (1985). Chemotactic activity of hydroxyalkenals on rat neutrophils. International Journal of Tissue Reactions, 7: 137-142.
- 11. Lee KS, Buck M, Houglum K et al. (1995). Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. Journal of Clinical Investigation, 96: 2461-2468.
- 12. Estabrook RW (1967). Mitochondrial respiratory control and the polarographic measurement of ADP:O ratio. In: Estabrook RW & Pullman ME (Editors), Methods in Enzymology Academic Press, New York, 41-47.
- 13. Teramoto K, Bowers JL, Khettry U et al. (1993). A rat fatty liver transplant model. Transplantation, 55: 737-741.
- 14. Willumsen N, Skorve J & Hexeberg S (1993). The hypotriglyceridemic effect of eicosapentaenoic acid in rats is reflected in increased mitochondrial fatty acid oxidation followed by dimished lipogenesis. Lipids, 28: 683-690.
- 15. Sekiya M, Yahagi N & Matsuzaka T (2003). Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology, 38: 1529-1539.
- 16. Clarke SD & Jump DB (1994). Dietary polyunsaturated fatty acid regulation of gene transcription. Annual Review of Nutrition, 14: 83-98.
- 17. Letteron P, Fromenty B, Terris B et al. (1996). Acute and chronic hepatic steatosis leads to in vivo lipid peroxidation in mice. Journal of Hepatology, 24: 200-208.
- 18. Oliveira CPMS, Gayotto LC, Tatai C et al. (2002). Oxidative stress in the pathogenesis of nonalcoholic fatty liver disease, in rats fed with a choline-deficient diet. Journal of Cellular and Molecular Medicine, 6: 399-406.
- 19. Oliveira CPMS, Gayotto LC, Tatai C et al. (2003). Vitamin C and vitamin E in prevention of nonalcoholic fatty liver disease (NAFLD) in choline deficient diet fed rats. Nutrition Journal, 2: 9.
- 20. Pessayre D, Mansouri A & Fromenty B (2002). Nonalcoholic steatosis and steatohepatitis: mitochondrial dysfunction in steatohepatitis. American Journal of Physiology, 282: G193-G199.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
04 May 2006 -
Date of issue
Feb 2006
History
-
Accepted
22 Sept 2005 -
Received
04 May 2005