Abstract
Carboxypeptidase M (CPM) is an extracellular glycosylphosphatidyl-inositol-anchored membrane glycoprotein, which removes the C-terminal basic residues, lysine and arginine, from peptides and proteins at neutral pH. CPM plays an important role in the control of peptide hormones and growth factor activity on the cell surface. The present study was carried out to clone and express human CPM in the yeast Pichia pastoris in order to evaluate the importance of this enzyme in physiological and pathological processes. The cDNA for the enzyme was amplified from total placental RNA by RT-PCR and cloned in the vector pPIC9, which uses the methanol oxidase promoter and drives the expression of high levels of heterologous proteins in P. pastoris. The cpm gene, after cloning and transfection, was integrated into the yeast genome, which produced the active protein. The recombinant protein was secreted into the medium and the enzymatic activity was measured using the fluorescent substrate dansyl-Ala-Arg. The enzyme was purified by a two-step protocol including gel filtration and ion-exchange chromatography, resulting in a 1753-fold purified active protein (16474 RFU mg protein-1 min-1). This purification protocol permitted us to obtain 410 mg of the purified protein per liter of fermentation medium. SDS-PAGE showed that recombinant CPM migrated as a single band with a molecular mass similar to that of native placental enzyme (62 kDa), suggesting that the expression of a glycosylated protein had occurred. These results demonstrate for the first time the establishment of a method using P. pastoris to express human CPM necessary to the development of specific antibodies and antagonists, and the analysis of the involvement of this peptidase in different physiological and pathological processes
Kinins; Human carboxypeptidase M; Recombinant protein; Pichia pastoris
Braz J Med Biol Res, February 2006, Volume 39(2) 211-217
High expression of human carboxypeptidase M in Pichia pastoris. Purification and partial characterization
R.B. Craveiro1, J.D. Ramalho3, J.R. Chagas3, P.H.M. Wang2, D.E. Casarini2, J.L. Pesquero3,4, R.C. Araújo3 and  Correspondence and Footnotes
Correspondence and Footnotes
 J.B. Pesquero1
J.B. Pesquero1
1Departamento de Biofísica, 2Departamento de Nefrologia, Escola Paulista de Medicina, Universidade Federal de São Paulo, São Paulo, SP, Brasil
3Universidade de Mogi das Cruzes, São Paulo, SP, Brasil
4Departamento de Fisiologia e Biofísica, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Carboxypeptidase M (CPM) is an extracellular glycosylphosphatidyl-inositol-anchored membrane glycoprotein, which removes the C-terminal basic residues, lysine and arginine, from peptides and proteins at neutral pH. CPM plays an important role in the control of peptide hormones and growth factor activity on the cell surface. The present study was carried out to clone and express human CPM in the yeast Pichia pastoris in order to evaluate the importance of this enzyme in physiological and pathological processes. The cDNA for the enzyme was amplified from total placental RNA by RT-PCR and cloned in the vector pPIC9, which uses the methanol oxidase promoter and drives the expression of high levels of heterologous proteins in P. pastoris. The cpm gene, after cloning and transfection, was integrated into the yeast genome, which produced the active protein. The recombinant protein was secreted into the medium and the enzymatic activity was measured using the fluorescent substrate dansyl-Ala-Arg. The enzyme was purified by a two-step protocol including gel filtration and ion-exchange chromatography, resulting in a 1753-fold purified active protein (16474 RFU mg protein-1 min-1). This purification protocol permitted us to obtain 410 mg of the purified protein per liter of fermentation medium. SDS-PAGE showed that recombinant CPM migrated as a single band with a molecular mass similar to that of native placental enzyme (62 kDa), suggesting that the expression of a glycosylated protein had occurred. These results demonstrate for the first time the establishment of a method using P. pastoris to express human CPM necessary to the development of specific antibodies and antagonists, and the analysis of the involvement of this peptidase in different physiological and pathological processes
Key words: Kinins, Human carboxypeptidase M, Recombinant protein, Pichia pastoris
Introduction
Carboxypeptidase M (CPM) is an extracellular glycosylphosphatidyl-inositol-anchored membrane glycoprotein. This protein is a member of the CPN/E subfamily of zinc metallo-carboxypeptidase. It specifically removes C-terminal basic residues such as lysine and arginine from peptides and from proteins at neutral pH (1-4). The highest levels of CPM have been found in human lung and placenta, but significant amounts are present in kidney, blood vessels, intestine, brain, and peripheral nerves (1,5-7). CPM has been found also in soluble form in various body fluids, including amniotic fluid, seminal plasma and urine (5,8,9). Due to its wide distribution in a variety of tissues, it is believed to play important roles in the control of peptide hormone and growth factor activity on the cell surface, and in the membrane-localized degradation of extracellular proteins (10).
By removing the C-terminal Arg residue of the intact kinins (and related peptides), CPM can switch the receptor specificity (11), inactivating the agonist for the B2 receptor and concomitantly producing an agonist for the B1 receptor (1). Recent results from our group have shown that the kinin B1 receptor is important for neutrophil migration into inflamed tissues (12,13) and for glucose/insulin homeostasis (Araujo RC, unpublished data). Furthermore, CPM is recognized by antibodies that detect cell surface antigens on macrophages and is strongly induced in the differentiation of monocytes to macrophages (14). Therefore, the mechanism for neutrophil migration could be dependent on the CPM present in the cell membrane of macrophages, indicating a relevant role of this enzyme at sites of inflammation. In addition, the conversion of agonistic activity was shown to result in an enhanced and prolonged output of nitric oxide in response to bradykinin or kallidin in cytokine-stimulated human lung microvascular endothelial cells (15). By cloning the cDNA from different human tissues, we could determine the molecular structure (16) and show the presence of alternatively spliced messages for CPM. Furthermore, we could also show that the expression of this carboxypeptidase in tumor tissues is differently regulated, raising the question of the role of this enzyme in cancer (16). Thus, taking into account the function played by kinins and nitric oxide in inflammatory diseases and cancer (11), and the fact that specific inhibitors of this carboxypeptidase are still lacking, CPM might be an important target for the development of new drugs to treat these pathologies.
Recently, a human CPM (hCPM) was expressed by using a system based on baculovirus-infected insect cells (Sf9) (17). The cited study aimed to identify the membrane-anchor signal and the glycosylphosphatidyl-inositol attachment site and to investigate the roles of the two conserved glutamic acid residues, Glu260 and Glu264, in kinetic parameters and in protein stability. Furthermore, the same group determined the crystal structure of the human enzyme (18).
Therefore, the objective of the present study was to obtain an active glycosylated protein using the expression system of the yeast Pichia pastoris. This system has several advantages, including the use of the alcohol oxidase I gene promoter, the ability to stably integrate expression plasmids at specific sites, the ability of the cells to be cultivated at high density, and a simplified purification procedure for secreted heterologous proteins. Moreover, similar to mammalian and insect cells, P. pastoris can carry out certain co- and post-translational modifications of foreign proteins (19,20). The results described in the present report demonstrate a high level of expression of the active recombinant hCPM with a molecular mass similar to the native glycoprotein (62 kDa), indicating that the P. pastoris expression system can be useful for the production of active CPM at relevant levels.
Material and Methods
Enzymes, primers and vectors for molecular biology procedures were purchased from Invitrogen (Carlsbad, CA, USA) and Promega (Madison, WI, USA). The dansyl-Ala-Arg substrate was from Bachem (King of Prussia, PA, USA). DNA sequencing was performed using an ABI377 sequencer (Applied Biosystems, Foster City, CA, USA). The Amicon Ultra 30,000 MWCO membrane was from Millipore (Billerica, MA, USA), and the Superdex 200 HR 10/30 and Mono Q HR 5/5 columns were from Amersham Biosciences (Piscataway, NJ, USA).
RNA extraction and reverse transcription polymerase chain reaction
Total RNA from human placenta was isolated using the Trizol reagent according to the manufacturer's protocol and its purity was evaluated by electrophoresis on 1% agarose gel. Contamination of RNA samples with genomic DNA was avoided by treatment for 1 h at 37ºC with 1 U RNAse-free/DNAse I per 2 µg RNA. For the same amount of RNA, the reaction also contained 20 U RnaseOUT-Rnase inhibitor and 3 mM MgCl2. After incubation, samples were heated to 95ºC and immediately chilled on ice for DNAse I denaturation. Reverse transcription was performed using 2 µg total RNA, 200 U Moloney murine leukemia virus II reverse transcriptase, 5 mM DTT, 50 ng random hexamer primers, 1X PCR buffer, 0.5 mM dNTPs, and 3 mM MgCl2. Reactions were carried out at 20ºC for 10 min, 42ºC for 45 min, 95ºC for 5 min, and 4ºC for 10 min. The resulting cDNA from the human placenta was then used for PCR which was performed with 500 ng cDNA. The PCR mixture contained 0.5 µM concentrations of sense (hCPM5 5'-GAATTCTTGCTGCCTT TGGTAGCTGC) and antisense (CPMAS2 5'-GCGGCCGCTTATTTGAAGAATAT GTGC) primers, which contained added sites for the restriction enzymes EcoRI and NotI, respectively, 2 U platinum TaqDNA polymerase, 1.5 mM MgCl2, 50 mM KCl, and 20 mM Tris-HCl, pH 8.3, in a 50-µL volume. PCR amplification was performed by incubating the samples at 94ºC for 1 min, followed by 30 cycles at 94ºC for 1 min, and at 60ºC for 90 s, with a final extension at 72ºC for 7 min. At the end of amplification, samples were submitted to electrophoresis on 1% agarose gel with a 100-bp DNA ladder as a size marker. The amplified bands of 1313 bp were visualized by ethidium bromide staining.
Construction of expression vector pPIC9-cpm
The PCR-amplified fragment encoding hCPM was cloned into the pGEM-T-Easy (Promega) cloning vector and subjected to double-stranded DNA sequencing (ABI 377, Applied Biosystems) After EcoRI-NotI digestion, the cpm gene was cloned in frame into the pPIC9 (Invitrogen) vector between the EcoRI (5' end) and NotI (3' end) restriction sites to generate the plasmid pPIC9-cpm.
Transformation of Pichia pastoris and selection of high-level expression colonies
The following culture media used for transformation of P. pastoris, selection of recombinant clones, and expression of CPM were prepared according to manufacturer recommendations (Invitrogen): minimal dextrose medium (MD), minimal methanol medium (MM), buffered glycerol-complex medium (BMGY), and buffered minimal methanol (BMMY). For cultures in liquid BMMY, which contain methanol as an inducer and carbon source, methanol was added every 24 h to a final concentration of 0.5% (v/v). All cultures were carried out at 30ºC. The pPIC9-cpm construct (10 µg) was linearized with BglII and transformed into P. pastoris wild-type strain GS115 (Invitrogen) by electroporation at 25 µF, 400 W, and 1500 V using a Bio-Rad (Hercules, CA, USA) GenePulser. Immediately after pulsing, 1 mL cold 1 M sorbitol was added to the P. pastoris cells, and plated onto MD containing 1 M sorbitol. After cultivation, all transformants were patched or replica plated onto both MM and MD plates to select the colonies growing more slowly in MM than in MD, and these colonies were cultivated in liquid BMMY. To analyze cpm gene integration into the P. pastoris genome, genomic DNA was isolated from different clones and integration was confirmed by PCR using the same cloning primers. CPM activity of positive clones in the BMMY supernatant was measured with the dansyl-Ala-Arg substrate as previously described (21). The clone presenting the highest CPM activity was inoculated into 100 mL BMGY and cultured for 1 day. The cells were then collected by centrifugation and resuspended in 500 mL BMMY. Every day (0, 24, 48, 72, and 96 h), just before the addition of 0.5% methanol, an aliquot of the medium was collected to measure CPM activity and protein concentration.
Purification of recombinant carboxypeptidase M
The CPM secreted in the supernatant was purified by a two-step procedure consisting of gel filtration and ion-exchange chromatography. Briefly, the expression medium was concentrated on an Amicon concentrator with an Amicon Ultra 30,000 MWCO membrane (Millipore), and loaded on a Superdex 200 HR 10/30 column (Amersham Biosciences) equilibrated with 50 mM Na-phosphate buffer, pH 7, containing 0.15 M NaCl. The column was eluted with the equilibration buffer at a flow rate 0.8 mL/min, and 0.5-mL fractions were collected and assayed for CPM activity. The effluent from the gel filtration column containing enzyme activity was combined and loaded onto a Mono Q HR 5/5 column (Amersham Biosciences) equilibrated with 50 mM Tris-HCl buffer, pH 8. The column was washed with equilibration buffer and then eluted with a linear gradient of 0 to 1 M NaCl in the same buffer. Proteins were detected by UV absorption at 280 nm, 0.5-mL fractions were collected and assayed for CPM activity.
Protein concentration was determined by the dye-binding method of Bradford (22) using bovine serum albumin as a standard.
Results and Discussion
Analysis of transformed clones and expression of human carboxypeptidase M
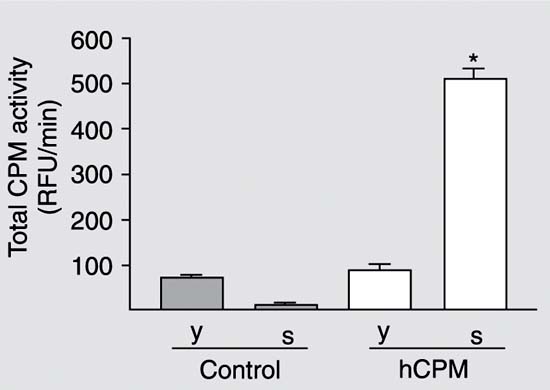
Total RNA from human placenta was isolated and submitted to the reverse transcription. The resultant cDNA was then used for PCR and yielded a 1313-bp DNA fragment containing the whole coding region with the expected sequence, except for one base change at position 1158 (T ® A; data not shown). However, this base alteration does not lead to a change in the amino acid sequence of the generated protein. After digestion with EcoRI-NotI restriction enzymes, the cpm gene was cloned in frame in the pPIC9. The generated plasmid pPIC9-cpm containing the hCPM gene was transformed into the P. pastoris wild-type strain GS115. Some of the colonies selected on MD plates (10 of 34 colonies) were tested by PCR to confirm the integration of the CPM coding region into the P. pastoris genome and six positive clones were selected. The clone with the highest activity (9.4 RFU min-1 mg protein-1) and one negative control, pPIC9 (without the insert), were initially inoculated into BMGY and later into BMMY. The cells and the supernatant of these clones were then collected by centrifugation, CPM activity was measured and protein concentration was determined. As shown in Figure 1, carboxypeptidase activity was detected at high levels in the supernatant of the positive clone. Figure 2 shows the levels of the total carboxypeptidase activity at different times. As observed, CPM activity increased in the medium in a time-dependent manner. The advantage of expressing heterologous proteins as secreted proteins is that P. pastoris secretes very low levels of native proteins. This, combined with the very low amount of protein in the minimal Pichia growth medium, means that the medium serves as the first step in the purification of the protein (19).
Expression of recombinant hCPM in the control and transformed (hCPM) clones of Pichia pastoris 24 h after inoculation into buffered glycerol-complex medium. The yeast cells (y) and the supernatant (s) were separated by centrifugation and assayed for CPM activity with dansil-Ala-Arg as substrate. hCPM = human carboxypeptidase M; RFU = relative fluorescence units. *P < 0.01 compared to the supernatant of the control clone (t-test).
Carboxypeptidase M activity in the culture supernatant of the GS115-CPM strain. Cells were induced in buffered minimal methanol and the activity was evaluated with the substrate dansil-Ala-Arg at different times. RFU = relative fluorescence units. *P < 0.05 and **P < 0.01 compared to the 24-h culture (t-test).
Purification of human carboxypeptidase M
The supernatant was concentrated through an Amicon membrane and submitted to gel filtration chromatography (Superdex 200 HR 10/30). Protein elution was monitored by measuring the absorbance at 280 nm and the carboxypeptidase activity was evaluated with the substrate dansyl-Ala-Arg. Protein eluted from the column showed one major peak of activity with a molecular mass estimated between 40 and 90 kDa (Figure 3A). The fractions corresponding to this major peak were pooled (P) and submitted to SDS-PAGE. The results showed the presence of three main bands in the gel (Figure 3B). The active fraction was submitted to ion-exchange chromatography (Mono Q 5/5) which resulted in only one peak of activity and a band in SDS-PAGE around 62-70 kDa (Figure 4), similar to the 62 kDa corresponding to the native placental protein. This small difference in molecular mass could be attributed to the partial cleavage produced by the P. pastoris STE13 protease in the recombinant protein, leaving four additional amino acids (EAEA) at the N-terminal region (23). Another reason could be the differences in the number and type of sugar units added to the yeast-secreted proteins compared with human proteins (24).
The purification steps resulted in 1753-fold purification of recombinant hCPM. The specific activity of the purified enzyme was 16474 RFU mg-1 min-1 using dansyl-Ala-Arg as substrate (Table 1). The very low levels of specific activity observed for the crude supernatant when compared to the purified enzyme may be explained by the presence of inhibitors of the enzyme in the fermentation medium. Some salts added to the medium at high concentrations are able to inhibit CPM and probably are separated from the enzyme during the purification process, causing a large increase in specific and total activities.
The latest achievements in the expression system of P. pastoris have changed deeply the methodology used to express heterologous proteins, increasing the expression levels and bioactivity of the latter. In addition, the great success of P. pastoris is related to the fact that this organism is a eukaryote, capable to express proteins with post-translational modifications, sometimes essential for proper function. A further benefit of the P. pastoris system is that strong promoters are available to drive the expression of the foreign gene(s), thus enabling production of large amounts of the target protein(s) with relative technical ease and at a lower cost than most other eukaryotic systems (25).
The results shown here demonstrate the establishment of a method using P. pastoris to express CPM in high yield of purified enzyme, i.e., 410 mg/L of fermentation culture. The expression levels of the hCPM in Pichia obtained in the present study was much higher (around 10-fold) when compared to the published expression system using baculovirus-infected insect cells, about 30-40 mg/L of recombinant mutant CPM in the culture medium (17). Some investigators, using the same system, have also described a similar recovery for the enzyme tannin acyl hydrolase (25), and even higher recoveries for the enzyme lipase/acyltransferase from Candida parapsilosis (EC 3.1.1.3) (26), and for the tetanus toxin fragment C (24,27), highlighting the potential of this technique to express high yield of heterologous proteins.
The present data show that we developed a methodology that permits the production of large amounts of the CPM enzyme, necessary for the development of specific antibodies and antagonists and for the analysis of the involvement of this peptidase in different physiological and pathological processes.
A, Gel filtration of recombinant human carboxypeptidase M (CPM). The concentrated supernatant (3.05 mg/mL in 2 mL) was loaded onto a Superdex 200 HR 10/30 column in the FPLC system. The column was equilibrated with 50 mM Na-phosphate buffer, pH 7, containing 0.15 M NaCl and eluted with the equilibration buffer at a flow rate of 0.8 mL/min. Fractions of 0.5 mL were collected and assayed for CPM activity. RFU = relative fluorescence units. B, SDS-PAGE (12%) of pooled fractions containing CPM activity (P). M, molecular mass markers (Bench Mark Protein Ladder, Invitrogen).
A, Ion-exchange chromatography. A pool of effluent containing enzyme activity (0.28 mg/mL in 1.5 mL) obtained by gel filtration (Figure 3) was loaded onto a Mono Q HR 5/5 column equilibrated with 50 mM Tris-HCl buffer, pH 8. The column was washed with equilibration buffer and then eluted with a linear gradient (0 to 1 M) NaCl in the same buffer. Proteins were detected by UV absorption at 280 nm. Fractions of 0.5 mL were collected and assayed for carboxypeptidase M activity. RFU = relative fluorescence units. B, SDS-PAGE (12%) of pooled active fractions. M, molecular mass markers (Bench Mark Protein Ladder, Invitrogen). Human carboxypeptidase M (hCPM) appears as a diffuse band because it is a glycoprotein.
Acknowledgments
We thank Nelson Mora and Ivan Cordeiro from the Departamento de Biofísica da Universidade Federal de São Paulo, São Paulo, SP, Brazil, for excellent technical assistance.
Research supported by FAPESP (Nos. 00/03850-5, 01/03409-0, 01/07538-9) and CNPq (No. 52.0012/02). R.B. Craveiro has a master degree fellowship from FAPESP (No. 02/04961-0). Received June 14, 2005. Accepted October 27, 2005.
- 1. Skidgel RA (1988). Basic carboxypeptidases: regulators of peptide hormone activity. Trends in Pharmacological Sciences, 9: 299-304.
- 2. Skidgel RA, McGwire GB & Li XY (1996). Membrane anchoring and release of carboxypeptidase M: Implications for extracellular hydrolysis of peptide hormones. Immunopharmacology, 32: 48-52.
- 3. Skidgel RA, Davis RM & Tan F (1989). Human carboxypeptidase M. Purification and characterization of a membrane bound carboxypeptidase that cleaves peptide hormones. Journal of Biological Chemistry, 264: 2236-2241.
- 4. Reznik SE & Fricker LD (2001). Carboxypeptidases from A to Z: implications in embryonic development and Wnt binding. Cellular and Molecular Life Sciences, 58: 1790-1804.
- 5. Skidgel RA (1996). Structure and function of mammalian zinc carboxy-peptidases. In: Hooper NM (Editor), Zinc Metalloproteases in Health and Disease Taylor and Francis Ltd., London, UK, 241-283.
- 6. Nagae A, Deddish PA, Becker RP et al. (1992). Carboxypeptidase M in brain and peripheral nerves. Journal of Neurochemistry, 59: 2201-2212.
- 7. Nagae A, Abe M, Becker RP et al. (1993). High concentration of carboxypeptidase M in lungs: presence of the enzyme in alveolar type I cells. American Journal of Respiratory Cell and Molecular Biology, 9: 221-229.
- 8. McGwire GB & Skidgel RA (1995). Extracellular conversion of epidermal growth factor (EGF) to [des-Arg53]EGF by carboxypeptidase M. Journal of Biological Chemistry, 270: 17154-17158.
- 9. Skidgel RA, Davis RM & Erdös EG (1984). Purification of a human urinary carboxypeptidase (kininase) distinct from carboxypeptidase A, B or N. Analytical Biochemistry, 140: 520-531.
- 10. Williams MC (2003). Alveolar type I cells: Molecular phenotype and development. Annual Review of Physiology, 65: 669-695.
- 11. Bhoola KD, Figueroa CD & Worthy K (1992). Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacological Reviews, 44: 1-80.
- 12. Pesquero JB, Araujo RC, Silva-Jr JA et al. (2000). Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proceedings of the National Academy of Sciences, USA, 97: 8140-8145.
- 13. Araujo RC, Kettritz R, Fichtner I et al. (2001). Altered neutrophil homeostasis in kinin B1 receptor-deficient mice. Biological Chemistry, 382: 91-95.
- 14. Krause SW, Rehli M & Andreesen R (1998). Carboxypeptidase M as a marker of macrophage maturation. Immunological Reviews, 161: 119-127.
- 15. Sangsree S, Brovkovych V, Minshall RD et al. (2003). Kininase I-type carboxypeptidases enhance nitric oxide production in endothelial cells by generating bradykinin B1 receptor agonist. American Journal of Physiology, 284: H1959-H1968.
- 16. Pessoa LG, Silva IDG, Baptista HA et al. (2002). Molecular structure and alternative splicing of the human carboxypeptidase M gene. Biological Chemistry, 383: 263-269.
- 17. Tan F, Balsitis S, Black JK et al. (2003). Effect of mutation of two critical glutamic acid residues on the activity and stability of human carboxypeptidase M and characterization of its signal for glycosylphosphatidylinositol anchoring. Biochemical Journal, 370: 567-578.
- 18. Reverter D, Maskos K, Tan F et al. (2004). Crystal structure of human carboxypeptidase M, a membrane-bound enzyme that regulates peptide hormone activity. Journal of Molecular Biology, 338: 257-269.
- 19. Sreekrishna K, Brankamp RG, Kropp KE et al. (1997). Strategies for optimal synthesis and secretion of heterologous protein in the methylotrophic yeast Pichia pastoris. Gene, 190: 55-62.
- 20. Cereghino JL & Cregg JM (2000). Heterologous protein expression in the methylotrophic yeast Pichia pastoris FEMS Microbiology Reviews, 24: 45-66.
- 21. Tan F, Deddish PA & Skidgel RA (1995). Human carboxypeptidase M. Methods in Enzymology, 248: 663-675.
- 22. Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72: 248-254.
- 23. Almeida MS, Cabral KS, Medeiros LN et al. (2001). cDNA cloning and heterologous expression of functional cysteine-rich antifungal protein Psd1 in the yeast Pichia pastoris. Archives of Biochemistry and Biophysics, 395: 199-207.
- 24. Cereghino GP, Cereghino JL, Ilgen C et al. (2002). Production of recombinant proteins in fermenter cultures of the yeast Pichia pastoris Current Opinion in Biotechnology, 13: 329-332.
- 25. Zhong X, Peng L, Zheng S et al. (2004). Secretion, purification, and characterization of a recombinant aspergillus oryzae tannase in Pichia pastoris Protein Expression and Purification, 36: 165-169.
- 26. Brunel L, Neugnot V, Landucci L et al. (2004). High-level expression of Candida parapsilosis lipase/acyltransferase in Pichia pastoris Journal of Biotechnology, 111: 41-50.
- 27. Clare J, Sreekrishna K & Romanos M (1988). Expression of tetanus toxin fragment C. Methods in Molecular Biology, 103: 193-208.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
04 May 2006 -
Date of issue
Feb 2006
History
-
Accepted
27 Oct 2005 -
Received
14 June 2005