Abstract
Gamma-irradiation (gamma-IR) is extensively used in the treatment of hormone-resistant prostate carcinoma. The objective of the present study was to investigate the effects of 60Co gamma-IR on the growth, cell cycle arrest and cell death of the human prostate cancer cell line DU 145. The viability of DU 145 cells was measured by the Trypan blue exclusion assay and the 3(4,5-dimethylthiazol-2-yl)-2,5,diphenyltetrazolium bromide test. Bromodeoxyuridine incorporation was used for the determination of cell proliferation. Cell cycle arrest and cell death were analyzed by flow cytometry. Superoxide dismutase (SOD), specifically CuZnSOD and MnSOD protein expression, after 10 Gy gamma-IR, was determined by Western immunoblotting analysis. gamma-IR treatment had a significant (P < 0.001) antiproliferative and cytotoxic effect on DU 145 cells. Both effects were time and dose dependent. Also, the dose of gamma-IR which inhibited DNA synthesis and cell proliferation by 50% was 9.7 Gy. Furthermore, gamma-IR induced cell cycle arrest in the G2/M phase and the percentage of cells in the G2/M phase was increased from 15% (control) to 49% (IR cells), with a nonsignificant induction of apoptosis. Treatment with 10 Gy gamma-IR for 24, 48, and 72 h stimulated CuZnSOD and MnSOD protein expression in a time-dependent manner, approximately by 3- to 3.5-fold. These data suggest that CuZnSOD and MnSOD enzymes may play an important role in the gamma-IR-induced changes in DU 145 cell growth, cell cycle arrest and cell death.
Gamma-irradiation; Prostate cancer; Proliferation; Cell death; Cell cycle arrest; Superoxide dismutase
Braz J Med Biol Res, February 2006, Volume 39(2) 227-236
Effects of gamma-radiation on cell growth, cycle arrest, death, and superoxide dismutase expression by DU 145 human prostate cancer cells
 Correspondence and Footnotes
Correspondence and Footnotes
 V. Vucic1, E.R. Isenovic2, M. Adzic1, S. Ruzdijic3 and M.B. Radojcic1
V. Vucic1, E.R. Isenovic2, M. Adzic1, S. Ruzdijic3 and M.B. Radojcic1
1Department of Molecular Biology and Endocrinology, 2Department of Radioisotopes,Vinca Institute of Nuclear Sciences, Belgrade, Serbia and Montenegro
3Laboratory of Molecular Neurobiology, Institute for Biological Research, Belgrade, Serbia and Montenegro
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Gamma-irradiation (g-IR) is extensively used in the treatment of hormone-resistant prostate carcinoma. The objective of the present study was to investigate the effects of 60Co g-IR on the growth, cell cycle arrest and cell death of the human prostate cancer cell line DU 145. The viability of DU 145 cells was measured by the Trypan blue exclusion assay and the 3(4,5-dimethylthiazol-2-yl)-2,5,diphenyltetrazolium bromide test. Bromodeoxyuridine incorporation was used for the determination of cell proliferation. Cell cycle arrest and cell death were analyzed by flow cytometry. Superoxide dismutase (SOD), specifically CuZnSOD and MnSOD protein expression, after 10 Gy g-IR, was determined by Western immunoblotting analysis. g-IR treatment had a significant (P < 0.001) antiproliferative and cytotoxic effect on DU 145 cells. Both effects were time and dose dependent. Also, the dose of g-IR which inhibited DNA synthesis and cell proliferation by 50% was 9.7 Gy. Furthermore, g-IR induced cell cycle arrest in the G2/M phase and the percentage of cells in the G2/M phase was increased from 15% (control) to 49% (IR cells), with a nonsignificant induction of apoptosis. Treatment with 10 Gy g-IR for 24, 48, and 72 h stimulated CuZnSOD and MnSOD protein expression in a time-dependent manner, approximately by 3- to 3.5-fold. These data suggest that CuZnSOD and MnSOD enzymes may play an important role in the g-IR-induced changes in DU 145 cell growth, cell cycle arrest and cell death.
Key words: Gamma-irradiation, Prostate cancer, Proliferation, Cell death, Cell cycle arrest, Superoxide dismutase
Introduction
Prostate cancer is the first in incidence and the second leading cause of cancer-related mortality in the human male population (1,2). Advanced disease is mostly treated by radiation therapy, sometimes in combination with hormone therapy or chemotherapy, but hormone withdrawal often leads to the selection of hormone-independent clones (3). The most desirable aim of radiotherapy is to efficiently eradicate tumor cells with minimal deleterious effects to the surrounding normal tissues and minimal consequent damage to the whole organism. Accordingly, induction of apoptotic cell death plays an important role (4-6). Radiation biology studies in rat prostate adenocarcinoma have shown that irradiation-induced apoptosis occurred at a higher rate in well-differentiated tumors compared to anaplastic ones (7). Thus, increased radiosensitivity, judged by apoptotic cell death as an end point, was observed for well-differentiated tumors (7). However, the results obtained for other systems demonstrated that gamma-irradiation (g-IR) is an inefficient inducer of apoptosis (8,9).
In addition to IR-induced cell killing, other clinically important effects of IR are related to cell proliferation and cell cycle arrest, i.e., to the control of tumor growth. Most normal cells exposed to IR characteristically activate cell cycle checkpoints, resulting in cell cycle arrest at the G1/S or G2/M checkpoints (10). Cancer cells are typically highly sensitive to radiation killing late in the G2 phase of the cell cycle (11). Also, when IR treatment with a high dose is used, the cell cycle checkpoints tend to be activated after exposure to IR, whereas for low dose IR this activation occurs during the radiation treatment. Thus, the effect of IR on the cell cycle should be defined as a function of time (11).
Radiation damage to cells invariably occurs via an increase in the local concentration of reactive oxygen species (ROS). The ROS increase also leads to induction of antioxidant defense mechanisms in the cell (12, 13). The first factor of enzymatic defense in the cell cytosol compartment is CuZn-superoxide dismutase (CuZnSOD), whereas its mitochondrial counterpart is MnSOD (13). On the basis of these findings, it appeared inviting to investigate the relationship between antioxidant enzymes and various g-IR-induced end points in cancer cells. Few data are available about carcinoma of the prostate, especially regarding the enzymatic antioxidant system. Only one or a few enzymes have been measured in normal and cancer prostatic tissue of men and rats, or in permanent cell lines (14-18). Therefore, in the present study we determined CuZnSOD and MnSOD protein expression in DU 145 cells exposed to g-IR in order to investigate whether these enzymes participate in g-IR-induced changes in DU 145 cell growth, cell proliferation, cell cycle, and cell death.
Material and Methods
Cell culture
The human prostate cancer cell line DU 145 was purchased from the American Type Culture Collection (Rockville, MD, USA). The cells were grown as monolayers in Eagle's medium supplemented with 10% heat-inactivated fetal calf serum, 100 IU/mL penicillin/streptomycin and 2 mM L-glutamine (Sigma Aldrich Chemie, Taufkirchen, Germany) at 37ºC under a 5% CO2 atmosphere.
Cell irradiation
Cell IR was performed during the exponential phase of cell growth, with 2, 10 or 20 Gy g-rays from a 60Co source (dose rate of 20 Gy/h).
Determination of cell growth and the cell viability index by the Trypan blue exclusion assay
For analysis of cell growth, DU 145 cells were plated onto 6-well plates (2 x 104 cells/well) and exposed to g-IR, and the Trypan blue exclusion (TBE) assay was performed 24, 48, and 72 h after g-IR as previously described (19). The viability index (VI) for each sample was calculated in relation to the corresponding control taken to be 100%.
Determination of cell proliferation (bromodeoxyuridine assay) and cell cytotoxicity (3(4,5-dimethylthiazol-2-yl)-2,5,diphenyltetrazolium bromide assay)
Following IR, g-IR and control cells were seeded in 96-well microtiter plates (5 x 103 cells/well). After 96 h, the cells were incubated with either the thymidine analogue bromodeoxyuridine (BrdU; Roche Diagnostic, Mannheim, Germany) or 3(4,5 dimethylthiazol-2-yl)-2,5,diphenyltetrazolium bromide (MTT; Sigma-Aldrich). The BrdU assay was performed according to the manufacturer's protocol, and the relative incorporation of BrdU was determined by measuring absorbance at 450 nm. After incubation with 20 µL of 1 mg/mL MTT for 4 h, the medium was removed, 100 µL of 0.1 M HCl in isopropanol was added and incubation was continued for an additional 1 h. Relative viability was determined by measuring absorbance at 550 nm.
Flow cytometry
For cell cycle analysis, the DU 145 cells were resuspended in 250 µL cold PBS and fixed with 0.5 mL ice-cold 70% ethanol. After the ethanol was washed out, the fixed cells were treated with 50 µg/mL RNAse A at 37ºC, incubated with 50 µg/mL propidium iodide (PI) at 4ºC for 30 min and then analyzed with a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Cell cycle distribution was determined using ModFIT software (Verity Software House, Inc., Topsham, ME, USA). In order to analyze cell death, approximately 1 x 105 DU 145 cells were mixed with 100 µL PBS containing 5 µL/mL annexin V-FITC and 5 µg/mL PI (Travigen Inc., Gaithersburg, MD, USA) before incubating at room temperature for an additional 15 min. After dilution with 400 µL of binding buffer, multiparameter measurements were performed. A minimum of 10,000 events were collected for each sample. The data were further processed with the LYSIS II software (Becton Dickinson).
Western immunoblotting analysis
Equal amounts of protein (20 µg) were separated on a 10% SDS-polyacrylamide gel and electrophoretically transferred to a nitrocellulose membrane (Schleicher and Schuell, Inc., Keene, NH, USA). Membranes were blocked in blocking buffer with 5% BSA in TBS-buffered saline (20 mM Tris, 137 mM NaCl, pH 7.6, containing 0.3% Tween 20), washed in TBS and incubated with the appropriate primary antibody against CuZnSOD, MnSOD and actin (1:3500; Stressgene Biotechnologies, Victoria, BC, Canada). The immunoblots were subsequently washed and incubated with anti-rabbit antibody (Oncogene Research Products, San Diego, CA, USA) covalently linked to alkaline phosphatase (1:4000) for 1 h. Signals were electronically digitalized by scanning and image intensity was quantitated with the ImageQuant software, version 1.2 (Molecular Dynamics, Sunnyvale, CA, USA).
Statistical analysis
Values are reported as means ± SEM, with N being the number of experiments. Statistical significance was evaluated by one-way (MTT, BrdU assay and Western blot) and two-way analysis of variance (ANOVA). If statistical significance was found, the Tukey post hoc test was used. The level of significance was set at P < 0.05.
Results
Effects of g -IR on DU 145 cell growth
The effects of g-IR on cell growth were analyzed by viable cell counts using the TBE assay, 24, 48, and 72 h after treatment with different doses of g-IR (2, 10, and 20 Gy). As the results show (Table 1), cell growth was significantly reduced after exposure to g-IR treatment and this reduction was time and dose dependent. The ratio between viable cell numbers in g-IR cells and control cells was reported as the VI (Table 1). We determined that the greatest effect of g-IR occurred at a dose of 20 Gy, which significantly decreased the viable cell number 72 h after treatment compared to control (control = 18.7 ± 0.6 x 105 cells/well; g-IR = 4.7 ± 0.4 x 105 cells/well, P < 0.001, N = 3). Thus, the calculated VI was 26%. g-IR treatment, also significantly decreased VI in a time- (F = 20, P < 0.001) and dose- (F = 32, P < 0.001) dependent fashion. In addition, the g-IR dose that induced a decrease of cell viability from 100 to 50% (IC50) was calculated to be 9.6 ± 0.9 Gy.
Time- and dose-dependent effects of g-irradiation on the viability index (VI) of DU 145cells.
To determine whether g-IR had a cytotoxic effect on DU 145 cells, we performed the MTT assay on DU 145 cells treated with g-IR (2, 10 and 20 Gy) and used for experiments 96 h after treatment (Figure 1). In these experiments the EC50 value, required to decrease viability from 100 to 50%, was not achieved. We determined that inhibition of cell viability was in the 14 to 45% range (for 2 and 20 Gy, respectively; Figure 1). Furthermore, when we increased the dose of g-IR to 30 Gy, the induced inhibition of cell viability was 46%. However, cell viability was significantly decreased in a dose-dependent manner (F = 38, P < 0.001, N = 3), with the maximum effect reached at a dose of 20 Gy (45 ± 1%, P < 0.001, N = 3).
We also determined whether or not g-IR had an antiproliferative effect on DU 145 cells using the BrdU incorporation assay. The g-IR dose of 20 Gy (Figure 2) inhibited DNA synthesis by 77 ± 2% (P < 0.001, N = 3). The ID50 value obtained was 9.7 ± 1.3 Gy, and this dose inhibited cell viability by 34 ± 2% as determined by the MTT assay, whereas the inhibition of g-IR DNA synthesis was statistically significant for all doses applied (F = 27, P < 0.001, N = 3).
Effect of different doses of gamma irradiation (g-IR) on DU 145 cell viability. Cells were exposed to different doses of g-IR (2, 10, and 20 Gy) and analyzed after 96 h. Cell viability was measured by the diphenyltetrazolium bromide assay. Untreated control samples were arbitrarily assigned a value of 100% and the results of all treatments were normalized to 100% (i.e., % of control). Data are reported as the mean ± SEM for three separate experiments performed in quadruplicate. ***P < 0.001 for g-IR cells vs control (one-way ANOVA followed by the Tukey post hoc test).
Cell proliferation after different doses of gamma irradiation (g-IR). DU 145 cells were exposed to different doses of g-IR (2, 10, and 20 Gy) and analyzed after 96 h. Cell proliferation was measured on the basis of bromodeoxyuridine incorporation during DNA synthesis. Untreated control samples were arbitrarily assigned a value of 100% and the results of all treatments were normalized to 100% (% of control). Data are reported as the mean ± SEM for three separate experiments performed in quadruplicate. ***P < 0.001 for g-IR cells vs control (one-way ANOVA followed by the Tukey post hoc test).
Cell cycle arrest and cell death under the influence of g -IR
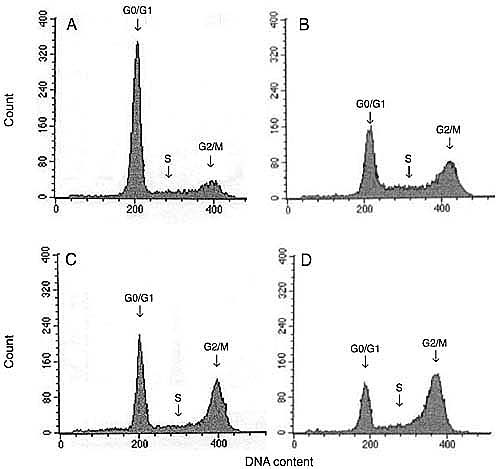
Since g-IR prevented cell growth, proliferation and DNA synthesis, we next used flow cytometry to test whether g-IR had an effect on cell cycle arrest. A gradual increase in the percentage of cells in the G2/M phase of the cell cycle occurred at 24, 48, and 72 h after a dose of 20 Gy (Figure 3). This increase was paralleled by a decrease in the cell population in the G0/G1 phase and was dose- (F = 844, P < 0.001, N = 2) and time- (F = 63, P < 0.001, N = 2) dependent. The changes in S phase only depended on the dose applied (F = 19, P < 0.001, N = 2), but not on the time after treatment (F = 3, p = 0.06, N = 2). In addition, the percentage of cells in the G2/M phase, 72 h after IR treatment (20-Gy dose) was significantly increased from 15.1 ± 2.0 (control cells) to 49.2 ± 1.3% (IR cells), whereas the number of cells in the G0/G1 phase was decreased from 71.2 ± 0.4 (control cells) to 29.1 ± 1.8% (IR cells; P < 0.001; Table 2).
The percentage of control cells in the subG0/G1 phase was low (8 ± 1%). No reproducible or consistent increase in the fraction of cells in the subG0/G1 phase was observed after g-IR. Furthermore, when the cells were treated for 24 h with 300 µM hydrogen peroxide, 100 µM etoposide, 1 µM doxorubicin, or UV light (50 J/m2), the percentage of cells in the subG0/G1 phase significantly increased, with the highest increase detected 48 h after treatment (data not shown). Thus, these cell treatments validated our method for analysis of the cell cycle.
Since previous results have indicated that g-IR inhibited cell growth, viability, proliferation, as well as cell cycle arrest, we tested the effect of g-IR on cell death. As shown in Table 3, g-IR significantly decreased viability in a time- and dose-dependent manner (F = 47 and F = 22, P < 0.001, N = 3), with the exception of the lowest dose, 2 Gy. The evidence for early apoptosis was minimal in both control and g-IR cells. Paralleling the increase in the radiation dose applied, the percentage of cells classified as early apoptotic increased from 0.3 ± 0.1 to 0.9 ± 0.2% (Table 3), but neither time- nor dose-dependent statistical significance was observed. Furthermore, the lack of apoptosis was confirmed by DNA gel electrophoresis, demonstrating no DNA ladder (data not shown). However, the radiation had dose- and time-dependent effects on the number of PI-positive cells in late apoptosis or necrosis (F = 20, P < 0.001; F = 54, P < 0.001, N = 3) and was not markedly increased after 2 Gy of IR, but was significantly increased after irradiation with 10 or 20 Gy (Table 3).
Representative flow cytometry histograms of the DU 145 cell cycle. A, Control cells. B, C, and D, 24, 48, and 72 h after treatment with 20 Gy gamma irradiation, respectively. The arrows indicate the phases of the cell cycle: G0/G1, S and G2/M.
Time- and dose-dependent effects of g-irradiation on the distribution of the DU 145 cell cycle.
Effects of g -IR on CuZnSOD and MnSOD protein expression in DU 145 cells
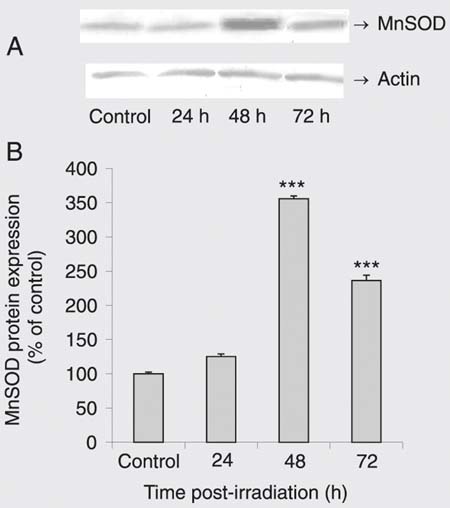
To determine whether g-IR induces SOD expression, we exposed DU 145 cells to 10 Gy g-IR for 24, 48, and 72 h. Western immunoblotting analysis with anti-CuZnSOD and MnSOD antibodies demonstrated that g-IR markedly enhanced both CuZnSOD (Figure 4) and MnSOD protein expression (Figure 5) in a time-dependent manner. No changes in loading (checked with anti-actin antibody) were detected.
Taken together, these data suggest that these SOD enzymes, CuZnSOD and MnSOD, may be involved in g-IR-mediated cell growth and proliferation.
Effect of gamma irradiation (g-IR) on the CuZn-superoxide dismutase (CuZnSOD) protein level. A, A representative Western blot. CuZnSOD behaves as a 16-kDa protein. Actin is indicated as a loading control. B, Results of densitometry analysis of separate Western blots (each bar is the mean ± SEM). ***P < 0.001 for g-IR cells vs control (one-way ANOVA followed by the Tukey post hoc test).
Effect of gamma irradiation (g-IR) on Mn-superoxide dismutase (MnSOD) protein level. A, A representative Western blot. MnSOD behaves as a 22-kDa protein. Actin is indicated as a loading control. B, Results of densitometry analysis of three separate Western blots (each bar is the mean ± SEM). ***P < 0.001 for g-IR cells vs control (one-way ANOVA followed by the Tukey post hoc test).
Discussion
We believe that our data on DU 145 cells demonstrate that 60Co g-IR is an effective anticancer agent leading to the induction of a G2/M cell cycle arrest and inhibition of proliferation of the hormone-refractory human prostate cancer cell line DU 145.
In the present study, we selected the doses of 2 and 10 Gy as representative of the 1.8- to 2-Gy daily clinical fractions given during curative radiotherapy and of the 8- to 10-Gy single doses given in palliative radiotherapy. We also applied a higher (20 Gy) dose that was in the range of the cumulative curative dose for carcinomas.
The data regarding viable cell counts (TBE assay) showed a significant decrease in the cell viability index, i.e., in the number of viable cells in g-IR DU 145 cells compared to control. These changes were dose and time dependent. This observation was confirmed by the MTT and BrdU assays. The inhibitory doses obtained in these experiments (EC50 and ID50) were significantly different (>20 and 9.7 Gy). The ID50 value was in accordance with the IC50 dose obtained from VI calculations (9.6 Gy). However, the EC50 dose was markedly higher than the ID50 dose. Thus, IR treatment within a clinically relevant dose range would predominantly express antiproliferative effects rather than cell killing. The significance of the IR-induced antiproliferative effects has been often underestimated compared to cell death (4,6-9), but in the present study we showed that a decrease of proliferation is the most prominent effect of g-IR on DU 145 cells.
Apoptotic cell death plays an important role in the death of both normal prostate and androgen-dependent malignant prostate tissue following androgen withdrawal, leading to a decrease in glandular or tumor volume, respectively. However, recent data support the hypothesis that apoptosis may not be the dominant mode of cell death following radio- and chemotherapy in stromal (i.e., fibroblast) and epithelial tissues (5,20). One mechanism by which cancer cells become resistant to IR or chemotherapy is disruption of the pathways that lead to apoptosis (21, 22). Alternately, permanent cell cycle arrest or senescence-like terminal growth arrest may also be factors in determining prostate cell death following radiotherapy.
Contrary to normal cells exposed to IR, which usually results in cell cycle arrest at the G1/S and G2/M checkpoints, cancer cells are typically sensitive to radiation killing in the late G2 phase of the cell cycle, especially if they have the mutant p53 gene (23). Depending on the extent of IR-induced damage, cells either repair the damage during cell cycle arrest and return to the normal cell cycle or die. Our results demonstrated significant dose- and time-dependent changes in the cell cycle. DU 145 cells exhibited a persistent G2/M arrest after exposure to IR. This correlates with a mutation in the p53 gene present in DU 145 cells (23). Radiation-induced accumulation of the cells in the G2/M phase of the cell cycle was most prominent 72 h after exposure to 20 Gy. The appearance of a subG0/G1 peak, representing apoptotic cells, was hardly detectable. Furthermore, it has been reported (24) that combined treatment with IR and genistein synergistically increased the radiosensitivity of DU 145 cells. Genistein plus IR could lead to more cells arrested in the G2/M phase, with a time-dependent increase in hyperdiploid cells and induction of apoptosis (24). Thus, the evaluation of potential signaling pathways having a role in IR regulation of the cell cycle will be further explored in our laboratory.
Changes of the enzymatic and nonenzymatic antioxidative systems in different human tissues due to malignant processes have been described (25-27). A correlation between the activity of free radicals and malignant changes is considered to be certain (25, 28). For example, free radical-induced DNA base modifications have been reported in cancerous tissues (29). It is possible that these changes may be important for the development of new therapeutical approaches (30). The antitumor effect of oxygen radicals has recently been proven (26). A reduced content of antioxidant enzymes in tumor tissue compared with normal tissue seems to be responsible for this effect (30). Overexpression of CuZnSOD correlated with increased IR resistance (31). Also, overexpression of MnSOD both at the mRNA and protein level was correlated with increased radiation resistance (32,33), whereas deficiency or a low level of the enzyme activity was associated with increased radiosensitivity (32,34). In our study we found significantly increased CuZnSOD and MnSOD expression in DU 145 cells after IR treatment. Based on the notion that IR affects biological systems by increasing the intracellular concentration of ROS, i.e., intracellular concentration of O2·- (12,35-37), it was expected that an in vitro IR challenge would result in the induction of CuZnSOD and MnSOD in DU 145 cells. Leach et al. (37) demonstrated increased ROS concentration in DU 145 cells after 1- to 10-Gy IR. In addition, treatment of DU 145 cells with the nucleoside analog, 8-Cl-cAMP, also induced a G2/M arrest with a poor induction of apoptosis, but with time-dependent stimulation of both CuZnSOD and MnSOD protein expression (data not shown). The increase in SODs may be the cause of the relatively high radiation resistance of DU 145 cells reflected in high IC50, EC50 and ID50 values. Also, it has been reported that MnSOD is often overexpressed in cancer tissues, especially in the presence of the mutant p53 gene (38). It is known that overexpression of human MnSOD substantially protects cells from IR injury (32) and this could be the main reason why DU 145 cells are unable to execute the apoptotic cell death program following IR, as observed in our results. In addition, as hydrogen peroxide is known to be an important mediator of cell cycle regulation (39) and as its overproduction may lead to cell cycle arrest in the G2 phase (40), the IR-induced SOD expression and G2/M arrest in DU 145 cells may be the crucial processes, linked via a similar hydrogen peroxide signaling mechanism.
Our study identifies some of the potential molecular mechanisms involved in the DU 145 cell response to g-IR. The results indicate that the specific response of DU 145 cells to g-IR may result from altered expression of CuZnSOD and MnSOD enzymes.
Research supported by grants from the Ministry for Science, Technology and Development of Serbia (BOI-1953 and BOI-1641). Received January 21, 2005. Accepted September 14, 2005.
- 1. Jemal A, Thomas A, Murray T et al. (2002). Cancer statistics. CA: A Cancer Journal for Clinicians, 52: 23-47.
- 2. Marinovic V, Nedic O, Stanojevic N et al. (2000). Investigation of the relationship between two major prostate tumor markers. Yugoslav Medical Biochemistry, 19: 407-410.
- 3. Raghavan D (1988). Non-hormone chemotherapy for prostate cancer: principles of treatment and application to the testing of new drugs. Seminars in Oncology, 15: 371-389.
- 4. Algan O, Stobbe CC, Helt M et al. (1996). Radiation inactivation of human prostate cancer cells: the role of apoptosis. Radiation Research, 146: 267-275.
- 5. Bromfield GP, Meng A, Warde P et al. (2003). Cell death in irradiated prostate epithelial cells: role of apoptotic and clonogenic cell kill. Prostate Cancer and Prostatic Diseases, 6: 73-85.
- 6. Geldof AA & Slotman BJ (1996). Radiosensitizing effect of cisplatin in prostate cancer cell lines. Cancer Letters, 101: 233-239.
- 7. Woynarowska BA, Roberts K, Woynarowski JM et al. (2000). Targetting apoptosis by hydroxymethylacylfulvene in combination with gamma radiation in prostate tumor cells. Radiation Research, 154: 429-438.
- 8. Li WX & Franklin WA (1998). Radiation and heat-induced apoptosis in PC-3 prostate cancer cells. Radiation Research, 150: 190-194.
- 9. Sklar GN, Eddy HA, Jacobs SC et al. (1993). Combined antitumor effect of surramin plus irradiation on human prostate cancer cells: The role of apoptosis. Journal of Urology, 150: 1526-1532.
- 10. Hartwell LH & Kastan MB (1994). Cell cycle control and cancer. Science, 266: 1821-1828.
- 11. Deweese TL, Shipman JM, Dillehay LE et al. (1998). Sensitivity of human prostatic carcinoma cell lines to low dose rate radiation exposure. Journal of Urology, 159: 591-598.
- 12. Ames BA, Shingenaga MK & Park EM (1991). Oxidative damage of macromolecules. In: Davis K (Editor), Oxidation Damage and Repair: Chemical, Biological and Medical Aspects. Pergamon Press, Elmsford, NY, USA, 181-187.
- 13. Mates JM, Perez-Gomez C & De Castro IN (1999). Antioxidant enzymes and human diseases. Clinical Biochemistry, 32: 595-603.
- 14. Tunn S, Nass R, Ekkernkamp A et al. (1989). Evaluation of average life span of epithelial and stromal cells of human prostate by superoxide dismutase activity. Prostate, 15: 263-271.
- 15. Dierickx PJ & Asnong CJ (1982). Prostatic glutathione S-transferase in rat, guinea pig and rabbit. Toxicology Letters, 13: 225-229.
- 16. Baker AM, Oberley LW & Cohen MB (1997). Expression of antioxidant enzymes in human prostatic adenocarcinoma. Prostate, 32: 229-233.
- 17. Oberley TD, Zhong W, Szweda LI et al. (2000). Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate, 44: 144-155.
- 18. Jung K, Seidel B, Rudolph B et al. (1997). Antioxidant enzymes in malignant prostate cell lines and in primary cultured prostatic cells. Free Radical Biology and Medicine, 23: 127-133.
- 19. Vucic V, Adzic M, Niciforovic A et al. (2004). Cell death in irradiated prostate cancer cells assessed by flow cytometry. Yugoslav Medical Biochemistry, 23: 343-350.
- 20. Olive PL, Vikse CM & Vanderbyl S (1999). Increase in the fraction of necrotic, not apoptotic, cells in SiHa xenograft tumors shortly after irradiation. Radiotherapy and Oncology, 50: 113-119.
- 21. Kyprianou N, King ED, Bradbury D et al. (1997). Bcl-2 overexpression delays radiation-induced apoptosis without affecting clonogenic survival in human prostate cancer cells. International Journal of Cancer, 70: 341-348.
- 22. Kimura K, Bowen C, Spiegel S et al. (1999). Tumor necrosis factor-a sensitizes prostate cancer cells to g-irradiation-induced apoptosis. Cancer Research, 59: 1606-1614.
- 23. Bohnke A, Westphal F, Schmidt A et al. (2004). Role of p53 mutations, protein function and DNA damage for the radiosensitivity of human tumor cells. International Journal of Radiation Biology, 80: 53-63.
- 24. Yan SX, Ejima Y, Sasaki R et al. (2004). Combination of genistein with ionizing radiation on androgen-independent prostate cancer cells. Asian Journal of Andrology, 6: 285-290.
- 25. Balasubramaniyan N, Subramanian S & Govindasamy S (1994). Status of antioxidant systems in human carcinoma of uterine cervix. Cancer Letters, 87: 187-192.
- 26. Cerutti PA (1994). Oxy-radicals and cancer. Lancet, 344: 862-863.
- 27. Beno I, Staruchova M, Volkovova K et al. (1995). Increased antioxidant enzyme activities in the colorectal adenoma and carcinoma. Neoplasma, 42: 265-269.
- 28. Sun Y (1990). Free radicals, antioxidant enzymes, and carcinogenesis. Free Radical Biology and Medicine, 8: 583-599.
- 29. Olinski R, Zastawny TH, Foksinski M et al. (1995). DNA base modifications and antioxidant enzyme activities in human benign prostatic hyperplasia. Free Radical Biology and Medicine, 18: 807-813.
- 30. Yoshikawa T, Kokura S, Tainaka K et al. (1995). A novel cancer therapy based on oxygen radicals. Cancer Research, 55: 1617-1620.
- 31. Kinnula VL & Crapo JD (2004). Superoxide dismutases in malignant cells and human tumors. Free Radical Biology and Medicine, 36: 718-744.
- 32. Sun J, Chen Y, Li M et al. (1998). Role of antioxidant enzymes on ionizing radiation resistance. Free Radical Biology and Medicine, 24: 586-593.
- 33. Motoori S, Majima HJ, Ebara M et al. (2001). Overexpression of mitochondrial manganese superoxide dismutase protects against radiation-induced cell death in the human hepatocellular carcinoma cell. Cancer Research, 61: 5382-5388.
- 34. Bravard A, Ageron-Blanc A, Alvarez S et al. (2002). Correlation between antioxidant status, tumorigenicity and radiosensitivity in sister rat cell lines. Carcinogenesis, 23: 705-711.
- 35. Droge W (2001). Free radicals in the physiological control of cell function. Physiological Reviews, 82: 47-95.
- 36. Starke-Reed PE & Oliver CN (1989). Protein oxidation and proteolysis during aging and oxidative stress. Archives of Biochemistry and Biophysics, 275: 559-567.
- 37. Leach JK, Van Tuyle G, Lin PS et al. (2001). Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Research, 61: 3894-3901.
- 38. Pani G, Bedogni B, Anzevino R et al. (2000). Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Research, 60: 4654-4660.
- 39. Chang TS, Jeong W, Choi SY et al. (2002). Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. Journal of Biological Chemistry, 277: 25370-25376.
- 40. Flattery-O'Brien J & Dawes IW (1998). Hydrogen peroxide causes RAD9-dependent cell cycle arrest in G2 in Saccharomyces cerevisiae whereas manadion causes G1 arrest independent of RAD9 function. Journal of Biological Chemistry, 273: 8564-8571.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
04 May 2006 -
Date of issue
Feb 2006
History
-
Accepted
14 Sept 2005 -
Received
21 Jan 2005