Abstract
The objective of the present study was to determine the frequency of the most common clinical features in patients with autosomal dominant polycystic kidney disease in a sample of the Brazilian population. The medical records of 92 patients with autosomal dominant polycystic kidney disease attended during the period from 1985 to 2003 were reviewed. The following data were recorded: age at diagnosis, gender, associated clinical manifestations, occurrence of stroke, age at loss of renal function (beginning of dialysis), and presence of a family history. The involvement of abdominal viscera was investigated by ultrasonography. Intracranial alterations were prospectively investigated by magnetic resonance angiography in 42 asymptomatic patients, and complemented with digital subtraction arteriography when indicated. Mean age at diagnosis was 35.1 ± 14.9 years, and mean serum creatinine at referral was 2.4 ± 2.8 mg/dL. The most frequent clinical manifestations during the disease were arterial hypertension (63.3%), lumbar pain (55.4%), an abdominal mass (47.8%), and urinary infection (35.8%). Loss of renal function occurred in 27 patients (mean age: 45.4 ± 9.5 years). The liver was the second organ most frequently affected (39.1%). Stroke occurred in 7.6% of the patients. Asymptomatic intracranial aneurysm was detected in 3 patients and arachnoid cysts in 3 other patients. In conclusion, the most common clinical features were lumbar pain, arterial hypertension, abdominal mass, and urinary infection, and the most serious complications were chronic renal failure and stroke. Both intracranial aneurysms and arachnoid cysts occurred in asymptomatic patients at a frequency of 7.14%.
Autosomal dominant polycystic kidney disease; Hepatic cyst; Intracranial aneurysm; Arachnoid cyst; End-stage renal disease
Braz J Med Biol Res, April 2006, Volume 39(4) 533-538
Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease
E.A. Romão1, M. Moysés Neto1, S.R. Teixeira1, V.F. Muglia2, O.M. Vieira-Neto1 and  Correspondence and Footnotes
Correspondence and Footnotes
 M. Dantas1
M. Dantas1
1Divisão de Nefrologia, 2Centro de Análises por Imagem, Departamento de Clínica Médica, Hospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil
 Patients and Methods
Patients and Methods
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
The objective of the present study was to determine the frequency of the most common clinical features in patients with autosomal dominant polycystic kidney disease in a sample of the Brazilian population. The medical records of 92 patients with autosomal dominant polycystic kidney disease attended during the period from 1985 to 2003 were reviewed. The following data were recorded: age at diagnosis, gender, associated clinical manifestations, occurrence of stroke, age at loss of renal function (beginning of dialysis), and presence of a family history. The involvement of abdominal viscera was investigated by ultrasonography. Intracranial alterations were prospectively investigated by magnetic resonance angiography in 42 asymptomatic patients, and complemented with digital subtraction arteriography when indicated. Mean age at diagnosis was 35.1 ± 14.9 years, and mean serum creatinine at referral was 2.4 ± 2.8 mg/dL. The most frequent clinical manifestations during the disease were arterial hypertension (63.3%), lumbar pain (55.4%), an abdominal mass (47.8%), and urinary infection (35.8%). Loss of renal function occurred in 27 patients (mean age: 45.4 ± 9.5 years). The liver was the second organ most frequently affected (39.1%). Stroke occurred in 7.6% of the patients. Asymptomatic intracranial aneurysm was detected in 3 patients and arachnoid cysts in 3 other patients. In conclusion, the most common clinical features were lumbar pain, arterial hypertension, abdominal mass, and urinary infection, and the most serious complications were chronic renal failure and stroke. Both intracranial aneurysms and arachnoid cysts occurred in asymptomatic patients at a frequency of 7.14%.
Key words: Autosomal dominant polycystic kidney disease, Hepatic cyst, Intracranial aneurysm, Arachnoid cyst, End-stage renal disease
Introduction
The autosomal dominant polycystic kidney disease (ADPKD) belongs to a group of inherited polycystic kidney diseases (1) and is one of the most common hereditary disorders. Gene PKD1 mutations, localized in chromosome 16p13.3, whose gene product is polycystin protein 1, result in ADPKD type I which is responsible for 85 to 90% of ADPKD cases (2). Gene PKD2 mutations, localized in chromosome 4q21-23, whose gene product is polycystin protein 2, result in ADPKD type II, corresponding to 10 to 15% of ADPKD cases (2). This disease has been characterized by the presence or bilateral multiple cysts in the kidneys, associated or not with cystic alterations in other systems (3). The prevalence of ADPKD has been estimated to be 1:400 to 1:1000 individuals in the general population (4). Among patients with end-stage renal disease (ESRD) the prevalence of ADPKD is approximately 5-10% (3). In Brazil, a study reported a prevalence of 7.6%, corresponding to 74 of 975 patients observed at 15 hemodialysis centers (5), and another study reported a prevalence of 3% (6).
The renal and extrarenal manifestations of the disease are quite variable (3), even in individuals of the same family. These manifestations depend on the organs involved as well as on the intensity of the condition. The most common abnormalities related to the kidneys are: arterial hypertension, abdominal mass, urinary tract infection, lumbar or abdominal pain, hematuria, nephrolithiasis, and renal failure. The liver is the second most affected organ, with hepatic cysts that cause hepatomegaly and abdominal pain, although they may exceptionally cause hepatic dysfunction (3). Cysts may also occur occasionally in the pancreas, ovaries and choroid plexus. Other extrarenal manifestations include intracranial aneurysms (ICA) and aortic aneurysms, arachnoid cysts, cerebral artery dolichoectasia, colonic diverticula, and cardiac abnormalities such as mitral-valve prolapse, mitral regurgitation, aortic insufficiency, and tricuspid regurgitation (3).
ICA is one of the extrarenal manifestations of highest concern due to the risk of rupture and subarachnoid hemorrhage. The prevalence of ICA in patients with ADPKD ranges from 4 to 12%, compared to a prevalence of 1% for the general population (7). The risk of ICA rupture may be five times more frequent in patients with ADPKD than in the general population (8).
As far as we know, there are no reports of the evaluation of clinical data in Brazilian ADPKD patients. Thus, the aims of this study were: a) to determine the prevalence of renal and extrarenal manifestations in Brazilian patients with ADPKD, and b) to investigate the occurrence and characteristics of events related to ADPKD in the central nervous system.
Patients and Methods

We retrospectively reviewed the medical charts of patients with ADPKD at the University Hospital, Faculty of Medicine of Ribeirão Preto, University of São Paulo, from January 1985 to December 2003. The diagnosis of ADPKD was established by renal ultrasonography according to the criteria of Ravine et al. (9). A positive family history and the presence of hepatic cysts were considered and added as additional diagnostic criteria. Patients who did not fulfill the Ravine criteria, patients with exophytic cysts, calcifications or other alterations suggestive of malignancy, with associated angiomyolipomas or tuberous sclerosis, or patients with acquired simple cysts were excluded.
The sample studied consisted of 92 patients, 34 males (37%) and 58 females (63%). Twenty-one patients (22.8%) fulfilled 3 diagnostic criteria, 51 patients (55.4%) fulfilled 2 criteria and 20 (21.8%) presented ultrasonographic changes as the only diagnostic criterion, but all patients presented countless and bilateral renal cysts associated with enlargement of both kidneys, which are characteristic of ADPKD. Ultrasonography that resulted in a diagnosis of ADPKD was performed due to a previous family history of the disease in 10 males (10.9%) and in 8 females (8.7%). A casual diagnosis of ADPKD was made in 14 male patients (41.1%) and 41 female patients (70.6%) on the occasion of ultrasonographic screening for nephrolithiasis, arterial hypertension, back or abdominal pain, and abdominal mass. Among the female patients, non-obstetric ultrasonographic screening resulted in a diagnosis of ADPKD in 52 (89.6%) cases and routine obstetrical screening resulted in a diagnosis of ADPKD in 6 cases (10.4%). According to the medical records, it was not possible to determine the reasons for abdominal ultrasonographic screening tests in 8 male and 14 female patients.
Age, gender, clinical manifestations related to ADPKD such as abdominal or back pain, abdominal mass, hematuria, urinary tract infection, nephrolithiasis, arterial hypertension, and personal and family history of stroke were recorded. The age on the occasion of progression to ESRD (characterized as the beginning of dialysis treatment) and a family history of ADPKD were also recorded, together with the reasons to perform ultrasonography that resulted in a diagnosis of ADPKD. Laboratory tests included the determination of serum creatinine on the occasion of referral and on the occasion of the last outpatient visit (or at the time of ESRD diagnosis). Ultrasonography reports were also reviewed in order to identify involvement of other abdominal viscera.
A subgroup of 42 patients free of central nervous system-related symptoms was investigated prospectively from October 2002 to April 2004 by magnetic resonance angiography (MRA) in order to determine the occurrence of intracranial alterations. MRA was performed by multisequence using a time of flight three-dimensional magnetic resonance arteriogram obtained with 1.5 T, Magnéton Vision, Siemens, Erlangen, Germany. When the MRA findings were equivocal, digital subtraction angiography was employed to elucidate the case using the Advant-X equipment (General Electric Medical Systems, Milwaukee, WI, USA).
Statistical analysis
Categorical variables were compared between groups by the Fisher exact test, and continuous variables were compared between groups by the unpaired Student t-test (age at diagnosis and age at ESRD) or the Mann-Whitney U-test (serum creatinine at referral), with the level of significance set at P < 0.05. The study was approved by the local Research Ethics Committee and informed written consent was obtained from patients who underwent diagnostic tests for this study.
Results
Age at the time of ADPKD diagnosis, serum creatinine at referral, age at the beginning of the dialysis program, number of patients with hepatic cysts, and other clinical data are presented in Table 1. The frequency of arterial hypertension and urinary tract infection, but not of serum creatinine at referral, was higher in female than in male patients. Hepatic cysts were detected in 36 patients (39.1%), also showing a higher frequency in female patients. No cysts were detected in any other abdominal viscera examined by ultrasonography. A positive family history was present in 61.9% of the cases. ESRD occurred in 27 patients at a mean age of 45.4 ± 9.5 years during the study period.
Stroke occurred in 7 patients (7.6%), two cases being characterized as a hemorrhagic stroke due to rupture of intracranial aneurysms, and 5 cases probably as ischemic stroke. Stroke was more frequent in male patients (P = 0.04) and did not show association with arterial hypertension in this group (P = 0.09).
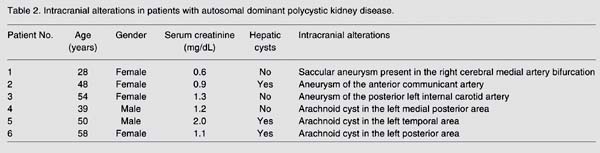
The search for intracranial alterations related to ADPKD was performed in 42 asymptomatic patients (18 males and 24 females). At the time of this investigation, mean age was 35.6 ± 14.3 years, and mean serum creatinine was 1.39 ± 0.79 mg/dL, and 3 of these patients were already on dialysis treatment. An ICA was detected by MRA in 5 patients, but after cerebral angiography it was confirmed in only 3 cases (7.1%), all of them female patients (Table 2). An arachnoid cyst was found in 3 patients (7.1%), one male and two females (Table 2). Twenty-nine patients (69.1%) showed normal cerebral MRA. Intracranial alterations considered to be nonspecific and not related to ADPKD (slight cerebral volume loss, presence of fetal circulation, left cerebellar venous drainage, old infarction, demyelinization or gliosis in 2 cases and one case of arteriolopathy associated with a hemorrhagic stroke) were found in 7 patients (16.7%).
Clinical and laboratory data of patients with autosomal dominant polycystic kidney disease.
Discussion
As far as we know, this is the first study that evaluated the renal and extrarenal clinical alterations of patients with ADPKD in a Brazilian population. The study showed a higher prevalence (63.0%) of the condition among women. Another study reported a 65.5% prevalence of ADPKD in American women (10), similar to this sample, while other investigators studying a Spanish population did not find any difference in ADPKD prevalence between genders (11).
Mean age at renal function loss was 47.9 years, similar to data reported by others for Spanish patients (11). Another study showed that mean patient age at the beginning of dialysis was 52.5 years among males and 58.1 years among females for German patients (12), suggesting a poorer outcome in male patients. However, another report showed that the mean age of patients reaching ESRD in Spain did not differ between genders (11), data similar to those reported in the present study. Also, in the present study, no significant difference was observed between genders in creatinine levels at referral, or in patient age when reaching dialysis, despite a higher frequency of arterial hypertension, presence of hepatic cysts and urinary tract infection in female patients. However, we would like to emphasize that arterial hypertension (13,14), younger age at diagnosis (13), mutations related to the PKD1 gene (13,15), and kidney size (16,17) have been identified as risk factors that might influence a faster progression to ESRD. In women, hepatic cystic disease is related to the earlier occurrence of renal insufficiency (3).
Other investigators have reported that the probability to be alive without reaching ESRD is 74% by the age of 50 years, 51% by the age of 58 years and 37% by the age of 70 years for Spanish patients (11). Other reports have stated that 57 (18) and 50% (19) of Canadian patients are alive without reaching ESRD by the age of 58 years. Other studies estimated that 45% of American and Canadian patients will have ESRD by the age of 60 years (13,19).
Lumbar pain and abdominal pain were the most frequent symptoms (55.4%) reported by the patients in the present study. In another study which evaluated pain patterns in American patients with polycystic kidney disease (20), back pain and abdominal pain were reported by 62.0 and 49.7% of the patients, respectively, after the diagnosis of polycystic kidney disease was established. Furthermore, pain was reported as the most frequent symptom in American patients that led to the diagnosis of polycystic kidney disease (20).
The prevalence of urinary infection was 35.8% (33 patients), mostly affecting female patients (87.8%; Table 1) in the present study. Symptomatic lower urinary tract infection affects 50-75% of all polycystic patients at some time.
Nephrolithiasis was observed in 16.3% of the patients (Table 1). It is an important manifestation of ADPKD, which occurs in approximately 20% among American patients (21). These frequencies exceed the estimated prevalence reported in a sample of the general American population, which varies from 4 to 9% in men, and from 1.7 to 4.1% in women (22).
Liver cysts were the only intra-abdominal cystic alterations found in the present investigation. The prevalence of liver cysts varies with age, being highly uncommon before the age of 16 years (23). Only 10% of American patients 20 to 29 years of age have liver cysts, whereas 75% of patients over the age of 60 have them (24). Although men and women have the same frequency of liver cysts, the number and size of cysts may be more severely increased in women (25).
Arterial hypertension was found in 63.3% of the present patients. The prevalence of arterial hypertension in American patients with ADPKD reported in other studies was approximately 60%, even with normal renal function (14), possibly affecting 80% of the ADPKD patients with renal failure (26). Another relevant issue related to arterial hypertension is stroke. In the present report, stroke was detected in 7 patients, with 2 of them being cases of confirmed hemorrhagic stroke.
Of the 42 patients evaluated by MRA in the present investigation, 3 (7.1%) showed ICA. A similar frequency was also obtained in three large prospective studies (4 to 11.7%) from USA (7,27,28). The presence of a positive family history is the main feature that affects the prevalence of ICA (29), reaching in these cases rates of 22 (7) and 25.8% (28). The frequency of ICA can also vary with age. ICA can also appear 10 years after a first examination that did not demonstrate the presence of this alteration, although at a low rate (2.6%) (30). Another American study reported that new ICA appeared after 6 to 33 years (mean of 14.2 years) in 5 of 20 patients with a previous demonstration of the presence of ICA (31). Some investigators have suggested that routine screening of all patients with ADPKD is not necessary because the risk of rupture is low (32). It has been suggested that this screening should be recommended only to patients with a positive family history, patients with a personal history of subarachnoid hemorrhage, patients who will be submitted to major surgeries, and to individuals with high-risk occupations (33).
Arachnoid cysts were found here in 3 patients (7.1%). In American patients, a prevalence rate of 8.1% was reported in one study (34) and of 5% in another (35), whereas this prevalence in the general population is approximately 1% (34,35). All of the 3 patients investigated here were asymptomatic, but the consequences of the presence of arachnoid cysts remain to be clarified (34,36).
Address for correspondence: M. Dantas, Divisão de Nefrologia, Departamento de Clínica Médica, HC, FMRP, USP, Av. Bandeirantes, 3900, 14048-900 Ribeirão Preto, SP, Brasil. Fax: +55-16-3633-6695. E-mail: mdantas@fmrp.usp.br
Research partially suported by Fundação de Assistência ao Ensino, Pesquisa e Assistência (FAEPA). Publication supported by FAPESP. Received July 18, 2005. Accepted February 7, 2006.
- 1. Wilson PD (2004). Polycystic kidney disease. New England Journal of Medicine, 350: 151-164.
- 2. Igarashi P & Somlo S (2002). Genetics and pathogenesis of polycystic kidney disease. Journal of the American Society of Nephrology, 13: 2384-2398.
- 3. Gabow PA (1993). Autosomal dominant polycystic kidney disease. New England Journal of Medicine, 329: 332-342.
- 4. Iglesias CG, Torres VE, Offord KP et al. (1983). Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. American Journal of Kidney Diseases, 2: 630-639.
- 5. Nunes A, Roisenberg I, Picolli E et al. (2003). Adult polycystic kidney disease in patients on haemodialysis in the south of Brazil. Nephrology, Dialysis, Transplantation, 18: 2686-2687.
- 6. Sesso R, Anção MS & Madeira SA (1994). Aspectos epidemiológicos do tratamento dialítico na grande São Paulo. Revista da Associação Médica Brasileira, 40: 10-14.
- 7. Huston 3rd J, Torres VE, Sulivan PP et al. (1993). Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 3: 1871-1877.
- 8. Rinkel GJ, Djibuti M, Algra A et al. (1998). Prevalence and risk of rupture of intracranial aneurysms: a systematic review. Stroke, 29: 251-256.
- 9. Ravine D, Gibson RN, Walker RG et al. (1994). Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet, 343: 824-827.
- 10. Schrier RW, McFann KK & Johnson AM (2003). Epidemiological study of kidney survival in autosomal dominant polycystic kidney disease. Kidney International, 63: 678-685.
- 11. Gonzalo A, Rivera M, Quereda C et al. (1990). Clinical features and prognosis of adult polycystic kidney disease. American Journal of Nephrology, 10: 470-474.
- 12. Gretz N, Zeier M, Geberth S et al. (1989). Is gender a determinant for evolution of renal failure? A study in autosomal dominant polycystic kidney disease. American Journal of Kidney Diseases, 14: 178-183.
- 13. Gabow PA, Johnson AM, Kaehny WD et al. (1992). Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney International, 41: 1311-1319.
- 14. Ecder T & Schrier RW (2001). Hypertension in autosomal-dominant polycystic kidney disease: early occurrence and unique aspects. Journal of the American Society of Nephrology, 12: 194-200.
- 15. Hateboer N, v Dijk MA, Bogdanova N et al. (1999). Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet, 353: 103-107.
- 16. Fick-Brosnahan GM, Tran ZV, Johnson AM et al. (2001). Progression of autosomal-dominant polycystic kidney disease in children. Kidney International, 59: 1654-1662.
- 17. Fick-Brosnahan GM, Belz MM, McFann KK et al. (2002). Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. American Journal of Kidney Diseases, 39: 1127-1134.
- 18. Churchill DN, Bear JC, Morgan J et al. (1984). Prognosis of adult onset polycystic kidney disease re-evaluated. Kidney International, 26: 190-193.
- 19. Parfrey PS, Bear JC, Morgan J et al. (1990). The diagnosis and prognosis of autosomal dominant polycystic kidney disease. New England Journal of Medicine, 323: 1085-1090.
- 20. Bajwa ZH, Sial KA, Malik AB et al. (2004). Pain patterns in patients with polycystic kidney disease. Kidney International, 66: 1561-1569.
- 21. Torres VE, Erickson SB, Smith LH et al. (1998). The association of nephrolithiasis and autosomal dominant polycystic kidney disease. American Journal of Kidney Diseases, 11: 318-325.
- 22. Soucie JM, Thun MJ, Coates RJ et al. (1994). Demographic and geographic variability of kidney stones in the United States. Kidney International, 46: 893-899.
- 23. Milutinovic J, Schabel SI & Ainsworth SK (1989). Autosomal dominant polycystic kidney disease with liver and pancreatic involvement in early childhood. American Journal of Kidney Diseases, 13: 340-344.
- 24. Milutinovic J, Fialkow PJ, Rudd TG et al. (1980). Liver cysts in patients with autosomal dominant polycystic kidney disease. American Journal of Medicine, 68: 741-744.
- 25. Gabow PA, Johnson AM, Kaehny WD et al. (1990). Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology, 11: 1033-1037.
- 26. Milutinovic J, Fialkow PJ, Agodoa LY et al. (1984). Autosomal dominant polycystic kidney disease: symptoms and clinical findings. Quarterly Journal of Medicine, 53: 511-522.
- 27. Chapman AB, Rubinstein D, Hughes R et al. (1992). Intracranial aneurysms in autosomal dominant polycystic kidney disease. New England Journal of Medicine, 327: 916-920.
- 28. Ruggieri PM, Poulos N, Masaryk TJ et al. (1994). Occult intracranial aneurysms in polycystic kidney disease: screening with MR angiography. Radiology, 191: 33-39.
- 29. Chauveau D, Pirson Y, Verellen-Dumoulin C et al. (1994). Intracranial aneurysms in autosomal dominant polycystic kidney disease. Kidney International, 45: 1140-1146.
- 30. Schrier RW, Belz MM, Johnson AM et al. (2004). Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. Journal of the American Society of Nephrology, 15: 1023-1028.
- 31. Belz MM, Fick-Brosnahan GM, Hughes RL et al. (2003). Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney International, 63: 1824-1830.
- 32. Gibbs GF, Huston 3rd J, Qian Q et al. (2004). Follow-up of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney International, 65: 1621-1627.
- 33. Perrone RD (1997). Extrarenal manifestations of ADPKD. Kidney International, 51: 2022-2036.
- 34. Schievink WI, Huston 3rd J, Torres VE et al. (1995). Intracranial cysts in autosomal dominant polycystic kidney disease. Journal of Neurosurgery, 83: 1004-1007.
- 35. Torres VE, Wiebers DO & Forbes GS (1990). Cranial computed tomography and magnetic resonance imaging in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 1: 84-90.
- 36. Wijdicks EF, Torres VE & Schievink WI (2000). Chronic subdural hematoma in autosomal dominant polycystic kidney disease. American Journal of Kidney Diseases, 35: 40-43.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
03 Apr 2006 -
Date of issue
Apr 2006
History
-
Accepted
07 Feb 2006 -
Received
18 July 2005