Abstract
Fabry disease is an X-linked lysosomal disorder due to a-galactosidase A deficiency that causes storage of globotriaosylceramide. The gene coding for this lysosomal enzyme is located on the long arm of the X chromosome, in region Xq21.33-Xq22. Disease progression leads to vascular disease secondary to involvement of kidney, heart and the central nervous system. Detection of female carriers based solely on enzyme assays is often inconclusive. Therefore, mutation analysis is a valuable tool for diagnosis and genetic counseling. Many mutations of the a-galactosidase A gene have been reported with high genetic heterogeneity, being most mutations private found in only one family. The disease is panethnic, and estimates of incidence range from about 1 in 40,000 to 60,000 males. Our objective was to describe the analysis of 6 male and 7 female individuals belonging to 4 different Fabry disease families by automated sequencing of the seven exons of the a-galactosidase gene. Sequencing was performed using PCR fragments for each exon amplified from DNA extracted from peripheral blood. Three known mutations and one previously described in another Brazilian family were detected. Of 7 female relatives studied, 4 were carriers. Although the present study confirms the heterogeneity of mutations in Fabry disease, the finding of the same mutation previously detected in another Fabry family from our region raises the possibility of some founder effect, or genetic drift. Finally, the present study highlights the importance of molecular analysis for carrier detection and genetic counseling.
Fabry disease; Lysosomal disorders; a-Galactosidase A; Globotriaosylceramide storage; GLA gene
Braz J Med Biol Res, December 2007, Volume 40(12) 1599-1604
Genomic analysis of Brazilian patients with Fabry disease
 Correspondence and Footnotes
Correspondence and Footnotes
 F.S. Pereira 1,2 , L.B. Jardim 3,4 , C.B. Netto 3 , M.G. Burin 3 , C. Cecchin 3 , R. Giugliani 1,2,3,5 *
*
These authors contributed equally to this study. Research supported by CAPES and FIPE-HCPA. Received June 1, 2006. Accepted April 18, 2007.
and U.S. Matte 1 *
*
These authors contributed equally to this study. Research supported by CAPES and FIPE-HCPA. Received June 1, 2006. Accepted April 18, 2007.
F.S. Pereira 1,2 , L.B. Jardim 3,4 , C.B. Netto 3 , M.G. Burin 3 , C. Cecchin 3 , R. Giugliani 1,2,3,5 *
*
These authors contributed equally to this study. Research supported by CAPES and FIPE-HCPA. Received June 1, 2006. Accepted April 18, 2007.
and U.S. Matte 1 *
*
These authors contributed equally to this study. Research supported by CAPES and FIPE-HCPA. Received June 1, 2006. Accepted April 18, 2007.
1 Centro de Terapia Gênica, 2 Programa de Pós-graduação em Clínica Médica, 3 Serviço de Genética Médica, 4 Serviço de Medicina Interna, Hospital de Clínicas de Porto Alegre, 5 Departamento de Genética, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil
 Patients and Methods
Patients and Methods
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Fabry disease is an X-linked lysosomal disorder due to a-galactosidase A deficiency that causes storage of globotriaosylceramide. The gene coding for this lysosomal enzyme is located on the long arm of the X chromosome, in region Xq21.33-Xq22. Disease progression leads to vascular disease secondary to involvement of kidney, heart and the central nervous system. Detection of female carriers based solely on enzyme assays is often inconclusive. Therefore, mutation analysis is a valuable tool for diagnosis and genetic counseling. Many mutations of the a-galactosidase A gene have been reported with high genetic heterogeneity, being most mutations private found in only one family. The disease is panethnic, and estimates of incidence range from about 1 in 40,000 to 60,000 males. Our objective was to describe the analysis of 6 male and 7 female individuals belonging to 4 different Fabry disease families by automated sequencing of the seven exons of the a-galactosidase gene. Sequencing was performed using PCR fragments for each exon amplified from DNA extracted from peripheral blood. Three known mutations and one previously described in another Brazilian family were detected. Of 7 female relatives studied, 4 were carriers. Although the present study confirms the heterogeneity of mutations in Fabry disease, the finding of the same mutation previously detected in another Fabry family from our region raises the possibility of some founder effect, or genetic drift. Finally, the present study highlights the importance of molecular analysis for carrier detection and genetic counseling.
Key words: Fabry disease, Lysosomal disorders, a-Galactosidase A, Globotriaosylceramide storage, GLA gene
Introduction
Fabry disease is an inborn error of metabolism of glycosphingolipids due to the deficiency of a-galactosidase A (a-Gal A; EC 3.2.1.22). The gene coding for this lysosomal enzyme is located on the long arm of the X chromosome, in region Xq21.33-Xq22 (1). It spans 12 kb and is divided into seven exons (2,3). The cDNA encodes a polypeptide of 429 amino acids including a 31-residue leader sequence. The a-Gal A enzyme is glycosylated and functions as a homodimer (4). Reduced a-Gal A activity causes storage of globotriaosylceramide inside cell lysosomes. Clinical manifestations include hypohidrosis, angiokeratomas, acroparesthesias, and corneal opacity. Disease progression leads to vascular disease secondary to involvement of kidney, heart and the central nervous system (5,6). "Cardiac" and "renal" variants present late-onset manifestations primarily limited to the heart (7) and kidney (renal variants), respectively (8).
Fabry disease is an X-linked disorder with male patients usually presenting classical symptoms although symptomatic females have also been described (9-11). The diagnosis of Fabry disease can be easily made in male patients by an enzymatic a-galactosidase (GLA) assay in plasma, leukocytes, or cultured fibroblasts but the carrier status of females is very difficult to determine (10). Due to random X inactivation, detection of female carriers based solely on enzyme assays is often inconclusive (12,13). Therefore, mutation analysis is a valuable tool for diagnosis and genetic counseling of affected families, especially regarding the availability of specific treatment, such as enzyme replacement therapy. However, no common mutations have been described in this gene. In the absence of a family member who has already received a diagnosis of the disorder, many cases are not diagnosed until adulthood (average age, 29 years) (14) when the pathology of the disorder may already be advanced. Although many mutations of the a-Gal A gene have been reported, most of them are based on genomic DNA sequences only (15). By 2005, more than 350 mutations had been described in the Human Gene Mutation Database (www.uwcm. ac.uk), including point mutations, small insertions/deletions, gross deletions, gross insertions and duplications, and complex rearrangements. Moreover, most mutations are found in one family, with a few exceptions (16). The rate of occurrence has been estimated at 1/40,000 or 1/60,000 men (5,11). Although Fabry disease is panethnic, very few reports on mutation analyses of Fabry disease in the Brazilian population are available in the literature.
Our service is a reference center for the diagnosis of lysosomal disorders in Brazil (17). In the present study, we describe the genomic analysis of 6 male Fabry patients belonging to 4 different families and their relatives.
Patients and Methods

Patients
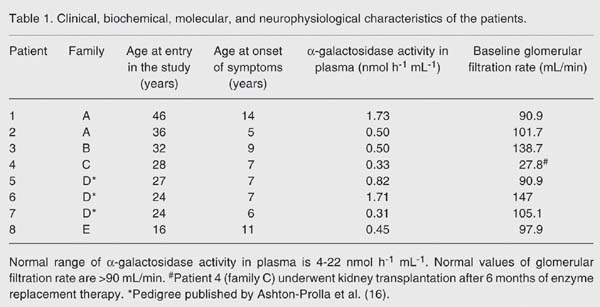
Four different families (named A, B, C, and E) were analyzed in this study, each with at least one affected male. All subjects had received a biochemical diagnosis based on the measurement of a-Gal A activity in leukocytes and/or in fibroblasts. Some of these patients are on enzyme replacement therapy with agalsidase a (patients 1, 2, 3, 4, 5, 6, and 7, belonging to families A, B, and C) or agalsidase ß (patient 8, family E) and have been described elsewhere (18,19).
A blood sample was collected in order to obtain nuclear DNA. After a pathogenic mutation was detected and confirmed in an individual, the result was communicated to the patient, and genomic studies were offered to their families.
The study was approved by the Ethics Research Committee of our Institution and by the Brazilian National Committee on Ethics in Research (CONEP) and the patients gave written informed consent.
PCR amplification
DNA was extracted from whole blood samples by the salting-out procedure (20). The genomic sequence of the seven exons of the GLA gene were amplified using the primers described by Ashton-Prolla et al. (16), except for exon 3 for which the reverse primer (F3-5'-GGCTCAGCTACCATGG CCT-3') was designed for this study. Reactions were carried out in a final volume of 50 µL, with 0.2 mM dNTP, 2.5 mM MgCl 2, 1X NH 4 buffer, 1 U Taq DNA polymerase, 20 pmol of each primer, and 100 ng DNA. All reagents were purchased from Invitrogen (Carlsbad, CA, USA). Reactions were performed in an Eppendorf personal cycler (Hamburg, Germany), with 30 cycles of 45 s and an annealing temperature of 47ºC for exon 4, 48ºC for exons 1, 2, 5, 6, and 7 and of 55ºC for exon 5, as shown in Table 1 . Amplification size ranged from 250 bp for exon 3 to 699 bp for exon 5.
Automated sequencing
PCR fragments were purified on Microspin S300 HR columns (Amersham, Little Chalfont, Buckinghamshire, England) according to manufacturer instructions prior to sequencing. Sequencing was performed with an ABI310 genetic analyzer using BigDye Terminator v3.1 (Applied Biosystems, Foster City, CA, USA). The same primers as used for PCR amplification were used for sequencing. Sequences were compared to the reference sequence NM000169 (www.ncbi.nih.gov) and any alterations were confirmed by reverse strand sequencing.
Results
Results
Four families with at least one affected member with Fabry disease were analyzed. Overall, 13 individuals were studied: 6 male patients as well as 7 possible carriers (mothers or sisters). The families were named A, B, C, and E, mostly because in previous local studies these were their designations and because another family, not included here, was called family D (19). The pathogenic mutation of family D has been already reported (16). Clinical and biochemical data are presented in Table 1 .
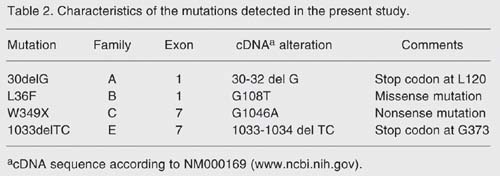
In three families, 3 different mutations that have been described were found: 30delG (16), W349X (21), and 1033delTC (21). In the fourth family, a novel mutation was detected in exon 1. One mutation (30delG, exon 1) had already been described in another Fabry kindred, called family D, living in our region (16). Table 2 summarizes the mutations found in the present study.
Genomic analysis of affected males
Mutation 30delG in exon 1 was found in family A with three affected males. As a result of the frameshift, a premature stop codon is introduced at residue 120. Mutation L36F was found in the only affected member of family B. This mutation changes a lysine codon (TTG) to a phenylalanine codon (TTT) in residue 36 but further studies are needed to clarify how this mutation may affect enzyme processing. Mutation W349X (21) found in family C is a nonsense mutation in exon 7. The protein, which should have 429 amino acids, presents a premature stop at residue 349, lacking its final 84 amino acids. Mutation 1033delTC, first described by Ashley et al. (21), found in family E, creates a stop codon 29 amino acids downstream from the deletion, at residue 373. The resulting protein, if successfully translated, would lack its final 56 amino acids.
Carrier detection
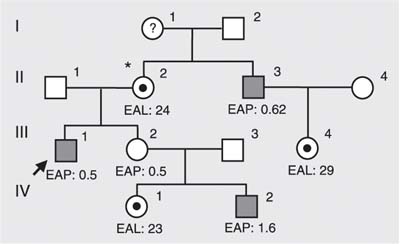
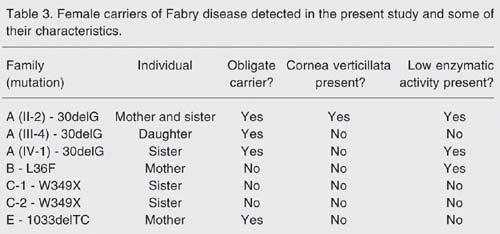
Three females from family A were studied and, as indicated by pedigree analysis (Figure 1 ), two could be considered obligate carriers: individual II.2 - mother and sister of affected males, and individual III-4, the daughter of an affected male. The third female (IV-1) is the sister of an affected male. All females were found to be carriers of the 30delG mutation. Enzyme activity was below reference values in II-2 and IV-1 and normal in III-4. Cornea verticillata was present in only one of them, II-2.
In family B, only one female, the mother of the affected patient, agreed to donate a sample for DNA analysis. This mother did not carry the L36F mutation, although her enzymatic activity was found to be below reference values. Her ophthalmologic evaluation was also normal. Patient B is, therefore, the carrier of a new mutation.
Two women, sisters of a male Fabry disease patient, were tested in family C. Both were asymptomatic, without cornea verticillata, and their enzymatic activity was within the control range. These women were found to be non-carriers of mutation W349X. The mother of both and of the affected male was not accessible.
Finally, the mother of the affected male of family E was a 1033delTC carrier. No clinical data were available and her enzymatic activity was within normal levels. These results are summarized in Table 3 .
Pedigree analysis of family A. All females were shown to be carriers of 30delG. *Woman with cornea verticillata. EAL = enzymatic activity in leukocytes (normal range: 26-53 nmol h-1 mg protein-1); EAP = enzymatic activity in plasma (normal range: 4-22 nmol h-1 mL-1). Individual III-2 was not available for mutation analysis. Hemizygote: filled squares. Heterozygote: circles with filled circles within.
Female carriers of the Fabry disease detected in the present study and some of their characteristics.
Clinical manifestations
The summary of informative findings is shown in Table 1 . There were two independent families carrying the same mutation -30delG: family A of the present paper, and the historical family D, already reported in the literature (16). Due to the small number of families in the present series, a search for genotype-phenotype correlations was limited to these two pedigrees. Both pedigrees segregating the 30delG mutation showed the highest levels of enzymatic activity, which could suggest an association between this type of mutation and some residual enzymatic activity.
Enzyme activity, age at diagnosis and glomerular filtration rate were used as parameters for clinical severity. As measured by renal involvement, the most severe mutation was W349X since this patient underwent kidney transplantation at age 25. The oldest subject in our sample, 36 years old (family A, subject 2, 30delG), although having multiple stroke-like episodes, still maintains its renal function preserved.
Regarding female relatives, clinical data were available only for the three carriers from family A (30delG). None of them has renal disease and only one was submitted to ophthalmologic exams, revealing the presence of cornea verticillata (II-2). As expected, enzyme activity did not correlate with genotype since it was below reference values in two carriers, normal in two others and low in one non-carrier mother.
Discussion
In the study we found four mutations, three of them (30delG, 1033delTC and W349X) previously described in other families affected with Fabry disease, and a novel mutation in exon 1 (L36F). It is generally assumed that Fabry mutations are private but this does not seem to be the case for our sample. Mutation 30delG had already been described in a large Brazilian family with Fabry disease (16). Apparently, these two families are not related but further studies using haplotype analysis shall clarify this question.
Seven female relatives were also studied and four were found to be carriers. Pedigree analysis and enzyme assay do not always provide enough information to determine carrier status, as observed in the present study. This illustrates the importance of determining the specific mutation in each family for precise heterozygote identification, prenatal diagnosis and genotype-phenotype correlation, although the last is still controversial (22). Additionally, carriers may present symptoms that could be amenable to treatment by enzyme replacement, but since these are not always specific, careful determination of carrier status is very important.
- 1. Kornreich R, Astrin KH, Desnick RJ. Amplification of human polymorphic sites in the X-chromosomal region q21.33 to q24: DXS17, DXS87, DXS287, and alpha-galactosidase A. Genomics 1992; 13: 70-74.
- 2. Kornreich R, Desnick RJ, Bishop DF. Nucleotide sequence of the human alpha-galactosidase A gene. Nucleic Acids Res 1989; 17: 3301-3302.
- 3. Eng CM, Niehaus DJ, Enriquez AL, Burgert TS, Ludman MD, Desnick RJ. Fabry disease: twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase A gene. Hum Mol Genet 1994; 3: 1795-1799.
- 4. Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab 2003; 80: 307-314.
- 5. Desnick RJ, Ioannou YA, Eng CM. a-Galactosidase A deficiency: Fabry disease. In: In Scriver CR, Beaudet AL, Sly WS, Valle D (Editors), The metabolic and molecular bases of inherited disease . 8th edn. New York: McGraw-Hill; 2001. p 3733-3774.
- 6. Pastores GM, Lien YH. Biochemical and molecular genetic basis of Fabry disease. J Am Soc Nephrol 2002; 13 (Suppl 2): S130-S133.
- 7. Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002; 105: 1407-1411.
- 8. Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int 2003; 64: 801-807.
- 9. MacDermot KD, Holmes A, Miners AH. Natural history of Fabry disease in affected males and obligate carrier females. J Inherit Metab Dis 2001; 24 (Suppl 2): 13-14.
- 10. Morrone A, Cavicchi C, Bardelli T, Antuzzi D, Parini R, Di Rocco M, et al. Fabry disease: molecular studies in Italian patients and X inactivation analysis in manifesting carriers. J Med Genet 2003; 40: e103.
- 11. Masson C, Cisse I, Simon V, Insalaco P, Audran M. Fabry disease: a review. Joint Bone Spine 2004; 71: 381-383.
- 12. Germain DP, Poenaru L. Fabry disease: identification of novel alpha-galactosidase A mutations and molecular carrier detection by use of fluorescent chemical cleavage of mismatches. Biochem Biophys Res Commun 1999; 257: 708-713.
- 13. Blaydon D, Hill J, Winchester B. Fabry disease: 20 novel GLA mutations in 35 families. Hum Mutat 2001; 18: 459.
- 14. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249-254.
- 15. Lai LW, Whitehair O, Wu MJ, O'Meara M, Lien YH. Analysis of splice-site mutations of the alpha-galactosidase A gene in Fabry disease. Clin Genet 2003; 63: 476-482.
- 16. Ashton-Prolla P, Ashley GA, Giugliani R, Pires RF, Desnick RJ, Eng CM. Fabry disease: comparison of enzymatic, linkage, and mutation analysis for carrier detection in a family with a novel mutation (30delG). Am J Med Genet 1999; 84: 420-424.
- 17. Coelho JC, Wajner M, Burin MG, Vargas CR, Giugliani R. Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. Eur J Pediatr 1997; 156: 650-654.
- 18. Gomes I, Nora DB, Becker J, Ehlers JA, Schwartz IV, Giugliani R, et al. Nerve conduction studies, electromyography and sympathetic skin response in Fabry's disease. J Neurol Sci 2003; 214: 21-25.
- 19. Jardim L, Vedolin L, Schwartz IV, Burin MG, Cecchin C, Kalakun L, et al. CNS involvement in Fabry disease: clinical and imaging studies before and after 12 months of enzyme replacement therapy. J Inherit Metab Dis 2004; 27: 229-240.
- 20. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
- 21. Ashley GA, Shabbeer J, Yasuda M, Eng CM, Desnick RJ. Fabry disease: twenty novel alpha-galactosidase A mutations causing the classical phenotype. J Hum Genet 2001; 46: 192-196.
- 22. Branton M, Schiffmann R, Kopp JB. Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol 2002; 13 (Suppl 2): S139-S143.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
29 Oct 2007 -
Date of issue
Dec 2007
History
-
Accepted
18 Apr 2007 -
Received
01 June 2006