Abstract
Iron is an essential metal for all living organisms. However, iron homeostasis needs to be tightly controlled since iron can mediate the production of reactive oxygen species, which can damage cell components and compromise the integrity and/or cause DNA mutations, ultimately leading to cancer. In eukaryotes, iron-regulatory protein 1 (IRP1) plays a central role in the control of intracellular iron homeostasis. This occurs by interaction of IRP1 with iron-responsive element regions at 5' of ferritin mRNA and 3' of transferrin mRNA which, respectively, represses translation and increases mRNA stability. We have expressed IRP1 using the plasmid pT7-His-hIRP1, which codifies for human IRP1 attached to an NH2-terminal 6-His tag. IRP1 was expressed in Escherichia coli using the strategy of co-expressing chaperonins GroES and GroEL, in order to circumvent inclusion body formation and increase the yield of soluble protein. The protein co-expressed with these chaperonins was obtained mostly in the soluble form, which greatly increased the efficiency of protein purification. Metal affinity and FPLC ion exchange chromatography were used in order to obtain highly purified IRP1. Purified protein was biologically active, as assessed by electrophoretic mobility shift assay, and could be converted to the cytoplasmic aconitase form. These results corroborate previous studies, which suggest the use of folding catalysts as a powerful strategy to increase protein solubility when expressing heterologous proteins in E. coli.
Iron metabolism; Iron-regulatory protein 1; Chaperonins; GroES; GroEL; Protein purification
Braz J Med Biol Res, April 2008, Volume 41(4) 270-276
Increased expression and purification of soluble iron-regulatory protein 1 from Escherichia coli co-expressing chaperonins GroES and GroEL
 Correspondence and Footnotes
Correspondence and Footnotes
 H. Carvalho1 and R. Meneghini2
H. Carvalho1 and R. Meneghini2
1Departamento de Ciências Biológicas, Universidade Federal de São Paulo, Diadema, SP, Brasil
2Centro Latino-Americano e do Caribe de Informações em Ciências da Saúde, São Paulo, SP, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Iron is an essential metal for all living organisms. However, iron homeostasis needs to be tightly controlled since iron can mediate the production of reactive oxygen species, which can damage cell components and compromise the integrity and/or cause DNA mutations, ultimately leading to cancer. In eukaryotes, iron-regulatory protein 1 (IRP1) plays a central role in the control of intracellular iron homeostasis. This occurs by interaction of IRP1 with iron-responsive element regions at 5' of ferritin mRNA and 3' of transferrin mRNA which, respectively, represses translation and increases mRNA stability. We have expressed IRP1 using the plasmid pT7-His-hIRP1, which codifies for human IRP1 attached to an NH2-terminal 6-His tag. IRP1 was expressed in Escherichia coli using the strategy of co-expressing chaperonins GroES and GroEL, in order to circumvent inclusion body formation and increase the yield of soluble protein. The protein co-expressed with these chaperonins was obtained mostly in the soluble form, which greatly increased the efficiency of protein purification. Metal affinity and FPLC ion exchange chromatography were used in order to obtain highly purified IRP1. Purified protein was biologically active, as assessed by electrophoretic mobility shift assay, and could be converted to the cytoplasmic aconitase form. These results corroborate previous studies, which suggest the use of folding catalysts as a powerful strategy to increase protein solubility when expressing heterologous proteins in E. coli.
Key words: Iron metabolism; Iron-regulatory protein 1; Chaperonins; GroES; GroEL; Protein purification
Introduction
Iron is an essential metal present as a prosthetic group in proteins such as hemoglobin, myoglobin, and cytochromes. However, it is also a potential source of reactive oxygen species (ROS) (1). Thus, iron levels need to be tightly controlled in order to keep a pool of "free" iron low enough not to cause cell damage. Iron is brought from the blood stream to the cells by transferrin, which binds to the transferrin receptor and is internalized into the cell. Inside the cells, vesicles containing iron-loaded transferrin/transferrin receptor are acidified, releasing iron, which is then used as a component of the prosthetic group for several proteins (2). Unused iron is stored by ferritin (3), preventing this metal from catalyzing ROS generation. The control of iron entry into the cell depends on the number of transferrin receptors, which are regulated by iron-regulatory protein (IRP) (4). IRP also regulates the synthesis of ferritin (5). When iron levels are low, transferrin receptor mRNA stability is increased by binding of IRP to iron-regulatory element (IRE) sequences at 3'-UTR. On the other hand, ferritin synthesis is inhibited by binding of IRP to IRE sequences at 5'-UTR of its mRNA, preventing protein translation (6-8). Two IRPs have been described: IRP1, which is more abundant and shows a mutually exclusive aconitase activity (6,9), and IRP2, which lacks this enzymatic activity (10). Binding of IRP1 to IRE sequences occurs when protein is devoid of a 4Fe-4S cluster as opposed to a situation in which the cluster and aconitase activity are present (11).
IRP1 is also activated in response to other stimuli. ROS (12-14) as well as reactive nitrogen species (15,16) have been shown to activate IRP1. It has also been shown that IRP1 can be phosphorylated and activated by protein kinase C (17), which makes iron-regulatory mechanisms even more complex.
Heterologous protein expression often needs to be addressed in many different ways in order to be successful. Escherichia coli is commonly used to produce recombinant proteins from several sources. Once protein is cloned and the expression vector is transferred to bacteria, expression conditions need to be optimized. These include shaker speed, temperature and absorbance (600 nm) used for expression induction (18,19). After all these efforts, the protein of interest may be produced in an insoluble form as inclusion bodies. Soluble protein recovery from these inclusion bodies is a difficult and often unsuccessful task. The use of molecular chaperonins to facilitate soluble protein expression has been adopted in some systems with success (20-24). Chaperonins are ubiquitous proteins that have an important role in the folding of newly synthesized proteins in vivo. The bacterial GroEL/GroES chaperonin complex is composed of two heptameric rings of GroEL and an attached heptameric lid of GroES, which assemble forming a "double doughnut" structure. Unfolded protein enters into this chamber and is folded in an ATP-dependent mechanism (25,26). It is estimated that 10-15% of all cytoplasmic proteins depend on GroEL/GroES under normal growth conditions in order to fold correctly (27). Under heat stress, this number increases to 30%.
We have expressed IRP1 in E. coli using a plasmid containing the human IRP1 sequence attached to an NH2-terminal 6-His tail. Expression of soluble IRP1 was only poor using this system. On the other hand, co-expression of IRP1 with chaperonins GroEL/GroES greatly improved the expression levels of soluble IRP1. This facilitated IRP1 purification, performed in two sequential steps: first with an Ni2+-Sepharose column and then by FPLC ion exchange chromatography. Protein purity was higher than 99%. IRP1 activity was confirmed by its ability to bind an IRE sequence in an electrophoretic mobility shift assay (EMSA).
Material and Methods
Material
Bromophenol blue, dithiothreitol, EDTA, HEPES, NP-40, heparin, ß-mercaptoethanol (EtSH), and Trizma were purchased from Sigma (USA). Acrylamide and bisacrylamide were from Bio-Rad (Hercules, USA). N,N,N',N'-tetramethylethylenodiamine was from Fluka (Buchs SG, Switzerland). a-[32P]-UTP was from GE Healthcare (USA). All other reagents were of analytical grade.
Plasmids
The plasmid pSPT-fer (4), kindly provided by Dr. Lukas Kühn (Lausanne, Switzerland), contains the IRE sequence for ferritin and the T7 and SP6 promoters. Plasmid pT7-His-hIRP1 was kindly provided by Dr. Mathias W. Hentze (Heidelberg, Germany). pT7-His-hIRP1 contains the sequence for human IRP1 fused to a 6-His tag, a T7 RNA polymerase promoter and an ampicillin selection marker.
IRP1 expression
IRP1 was expressed in BL21(DE3) E. coli strains transformed with plasmid pT7-His-hIRP1, previously transformed with plasmid pT-GroE, that codes for chaperonins GroEL and GroES. E. coli strains containing pT-GroE (20) were kindly provided by Dr. Shunsuke Ishii (University of Tsukuba, Japan). Colonies selected for both plasmids were inoculated into 5 mL Luria-Bertani medium containing 200 µg/mL carbenicillin and 20 µg/mL chloramphenicol and grown overnight at 37°C. This 5-mL culture was then inoculated into 500 mL Luria-Bertani medium containing 200 µg/mL carbenicillin and 20 µg/mL chloramphenicol and grown at 25°C, 150 rpm, until absorbance (600 nm) 0.9-1.0. Shaker speed was reduced to 80 rpm and protein expression was induced by the addition of 1 mM IPTG for 3 h. After this time, cells were centrifuged at 14,000 g, 4°C, for 10 min, and stored at -20°C until purification.
IRP1 purification
The bacterial pellet was defrosted, resuspended in lysis buffer (250 mM NaCl, 0.5% NP-40, 20 mM Tris-HCl, pH 8.0) and sonicated. Lysed bacteria were centrifuged at 14,000 g, 4°C, for 10 min. Purification was carried out in two steps. First, the supernatant was loaded onto a 2-mL Chelating Sepharose Fast Flow column (GE Healthcare), previously charged with Ni2+ ions, according to a modification of the protocol described by Gray et al. (28). Protein was loaded onto the column after adjusting the KCl concentration in the supernatant to 400 mM in order to inhibit non-specific binding. The column was washed with 8 mL washing buffer (WB, 24 mM HEPES, pH 7.6, 150 mM potassium acetate, 1.5 mM MgCl2, 5% glycerol) containing 400 mM KCl and then with 8 mL WB. Two other sequential washing steps were performed with 8 mL WB containing 10 and 20 mM imidazole. Elution was carried out with 10 mL WB containing 50 mM imidazole, with 2-mL fractions being collected. The column was further washed sequentially with WB containing 100 and 200 mM imidazole. The purity of the fractions was analyzed by SDS-PAGE and Coomassie blue staining and fractions containing higher amounts of IRP1 were pooled and loaded onto a MonoQ HR 5/5 ion exchange column coupled to an FPLC apparatus (GE Healthcare). Protein was eluted using a KCl gradient with a 4%/min slope and a flow rate of 1 mL/min. The purity of the fractions was analyzed by SDS-PAGE, Coomassie blue staining and densitometry. Quantification of the bands was done with the ImageQuant TL Software (GE Healthcare). Protein measurements were carried out with the Bradford assay (29).
Measurement of IRP1 activity by electrophoretic mobility shift assay
RNA probe and binding to IRP1. The 32P-labeled probe was synthesized using RiboProbe Gemini II from Promega (USA), plasmid pSPT-fer as a template and a-[32P]-UTP. The binding reaction was carried out as described (5). The following reagents were added to a 1.5-mL test tube: 2 µg purified IRP1 diluted in 4 µL Munro buffer (MB, 10 mM HEPES, pH 7.6, 3 mM MgCl2, 40 mM KCl, 5% glycerol, 1 mM DTT), 4 µL MB, with or without 4% EtSH (2% final concentration), 0.1 ng of the [32P]-RNA probe (15,000 cpm) in 4 µL MB, one unit RNase T1 in 4 µL MB (to degrade unprotected RNA, not bound to IRP1), 4 µL MB containing 50 mg/mL heparin and 4 µL of sample buffer (30 mM Tris-HCl, pH 7.5, 40% sucrose, 0.2% bromophenol blue). Each addition was followed by a brief spin to mix the reagents. EtSH, at the concentration used in the assay, fully activates IRP1 in vitro by reducing 4Fe-4S cluster sulfhydryl groups (30), which allows standardizing IRP1 activation as a function of the total amount of protein that can be activated.
Electrophoretic mobility shift assay. The method followed was essentially the one described by Konarska and Sharp (31). Samples were submitted to 6% acrylamide gel electrophoresis at 200 V. At the end of the run, the gel was dried in a Bio-Rad gel drier (USA), exposed for 24-48 h to a PhosphorImager Screen and read on a Storm PhosphorImager System (Molecular Dynamics, USA). Bands on the gel represent shifted bands of IRP1 bound to a 32P-labeled IRE-containing RNA probe. Results were analyzed with the ImageQuant Software (Molecular Dynamics).
Reassembly of 4Fe-4S cluster in vitro and conversion of IRP1 to cytosolic aconitase
The IRP1 4Fe-4S cluster was reassembled by the method of Gray et al. (28). Briefly, approximately 20 ng IRP1 was incubated with 10 mM cysteine and 50 µM FeSO4 for 30 min at 22°C. Conversion of IRP1 to cytosolic aconitase was indicated by the loss of IRP1 activity assayed by EMSA.
Results and Discussion
IRP1 plays a central role in iron homeostasis in eukaryotes, controlling the levels of the iron storage protein ferritin and the iron uptake protein transferrin receptor. This process is carried out by its binding to IRE sequences present in ferritin and transferring receptor mRNAs. In order to study the properties of recombinant IRP1, Gray et al. (28) and Brazzolotto et al. (32) expressed the protein in E. coli, recovering soluble IRP1. We made several attempts to express IRP1 in E. coli transformed with the same plasmid pT7-His-hIRP1, changing temperature, shaker speed and absorbance (600 nm) used for protein induction. However, we were not successful in these attempts, since most of the protein was produced in an insoluble form and we were unable to recover it by solubilization and renaturation. In an attempt to solve this problem, we decided to use folding catalysts to help expression of the protein in a soluble form, an approach that has been used by other investigators with success (20-24). The simultaneous expression of the bacterial chaperonins GroES/GroEL using the pT-GroE plasmid highly increased the expression of soluble IRP1, as shown in Figure 1. Optimal protein expression was achieved 90 min after induction with IPTG (Figure 2) and only a small amount of protein was present in the insoluble fraction.
IRP1 was then purified by metal affinity chromatography using an Ni2+-NTA column. Protein was loaded with a KCl concentration of 400 mM in order to minimize non-specific binding to the column. The column was washed with WB containing the same KCl concentration and then without KCl. The column was washed with WB containing 10-20 mM imidazole and protein was eluted with the same buffer containing 50 mM imidazole. In order to ensure that most of the protein was eluted with 50 mM imidazole, concentrations of 100 and 150 mM were also used to wash the column thereafter. Figure 3 shows that most IRP1 was eluted in the fractions containing 50 mM imidazole. The increase in IRP1 expression highly improved the efficiency of protein purification, since fractions that were much more enriched in IRP1 were obtained. Before the co-expression strategy, IRP1 was estimated to represent less than 10% of the total protein eluted with 50 mM imidazole. After the co-expression with chaperonins, IRP1 represented about 87% of the total protein eluted under the same conditions.
Following the first purification step, we used a second step of FPLC ion exchange chromatography to eliminate the contaminating bands shown in Figure 3, lanes 6-8. For this purpose, the fractions represented in these lanes, which were eluted with 50 mM and contained most IRP1, were loaded onto a MonoQ column. Elution was performed with a KCl gradient. As shown in Figure 4, IRP1 was eluted in fractions containing 200-300 mM KCl in high purity, estimated to be above 99% by gel densitometry.
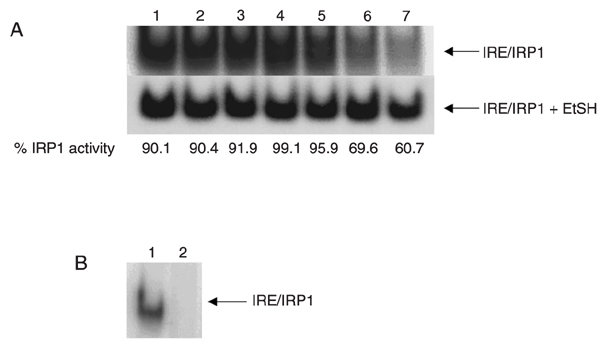
In order to test if IRP1 remained biologically active, purified protein was tested for its ability to bind an IRE-containing ferritin mRNA fragment. For this purpose, fractions obtained after ion exchange chromatography were assayed by EMSA. In this assay, total IRP1-binding activity is achieved by in vitro treatment with a potent reducing agent, ß-mercaptoethanol (EtSH) (30), which allows standardizing IRP1 activation as a function of the total amount of protein that can be activated. Figure 5A shows that fractions 5 through 11 from Figure 4, which correspond, respectively, to lanes 1-7 in Figure 5, present high IRE-binding activity, above 90% in most bands, compared to totally activated protein. In order to check if IRP1 could be converted to the cytosolic aconitase form, we treated purified IRP1 with 10 mM cysteine and 50 µM FeSO4, a protocol know to convert one form to the other, as described by Gray et al. (28). EMSA results are shown in Figure 5B. IRP1 treatment with FeSO4 and cysteine resulted in the abolition of IRP1 activity, probably due to the reassembly of the 4Fe-4S cluster and consequent conversion to cytosolic aconitase. Thus, apparently, neither co-expression of IRP1 with chaperonins nor the presence of the 6His-tag on the protein affected its ability to be converted to cytosolic aconitase.
The use of molecular chaperonins to solubilize expressed proteins has been described by others. Yasukawa et al. (20) described the effect of co-expressing chaperonins GroES and GroEL, as well as the redox active protein thioredoxin, along with vertebrate proteins in E. coli. All the eight proteins tested showed increased solubility when co-expressed with thioredoxin. On the other hand, co-expression of GroES and GroEL increased solubility of four of the proteins tested. Amrein et al. (21) described the effect of co-expressing GroES and GroEL in improving solubility of p50csk protein-tyrosine kinase. Mitsuda and Iwasaki (24) observed a great improvement in the expression of membrane-bound cytochrome P450 2B6 when co-expressed with GroES and GroEL in E. coli.
We observed a great improvement in the expression of soluble IRP1 co-expressed with chaperonins GroES and GroEL, which substantially facilitated the protein purification process. Our results, as well as results from other investigators, argue for the strategy of co-expressing chaperonins with the protein of interest in order to solve problems of protein solubility and expression yield when expressing heterologous proteins in E. coli. The crystal structure of IRP1 in the aconitase form (33) as well as the crystal structure of IRP1 complexed with ferritin IRE-RNA (34) were recently solved. However, the high purity of soluble active IRP1 obtained in this study may facilitate additional structural studies on IRP1.
Increase in the solubility of IRP1 by co-expression of chaperones GroES and GroEL. IRP1 was expressed in the BL21(DE3) Escherichia coli strain in the presence (+GroESL) or absence (-GroESL) of GroES and GroEL. IRP1 expression was induced or not with IPTG for 3 h and protein patterns for the soluble (s) and insoluble fractions (p) were analyzed by SDS-PAGE with Coomassie blue staining. IRP1 = iron-regulatory protein 1; i = samples induced with 1 mM IPTG; M = molecular weight marker.
Kinetics of IRP1 induction by IPTG for the bacterial clone that co-expresses IRP1 and GroEL. IRP1 was co-expressed in BL21(DE3) Escherichia coli strains expressing GroES and GroEL. IRP1 expression was induced with IPTG from 20 to 180 min, and protein patterns for the soluble (s) and insoluble fractions (p) were analyzed by SDS-PAGE with Coomassie blue staining. IRP1 = iron-regulatory protein 1; i = samples induced with 1 mM IPTG; M = molecular weight marker.
IRP1 purification using an Ni2+-NTA column. IRP1 was co-expressed in BL21(DE3) Escherichia coli strains expressing GroES and GroEL. IRP1 expression was induced with IPTG for 3 h, and soluble protein fractions were analyzed by SDS-PAGE with Coomassie blue staining. ft = flow-through; lanes 1,2 = wash with and without 400 mM KCl, respectively; lanes 3,4 = wash with 10 and 20 mM imidazole; lanes 5-9 = fractions eluted with 50 mM imidazole; lanes 10-14 = fractions eluted with 100 mM imidazole; lane 15 = fraction eluted with 150 mM imidazole. IRP1 = iron-regulatory protein 1. M = molecular weight marker.
IRP1 purification using ion exchange chromatography. Protein fractions 6-8, from Figure 3, were loaded onto a MonoQ column coupled to an FPLC apparatus. Elution was carried out with a KCl gradient until 1 M. The figure shows SDS-PAGE protein profiles stained with Coomassie blue. M = molecular weight marker; lane 1 = injected sample; lane 2 = fraction eluted before the salt gradient; lanes 3,4 = fractions eluted in 200 mM KCl; lanes 5-11 = fractions eluted between 200 and 300 mM KCl, containing purified IRP1; lane 12 = pooled fractions eluted between 300 and 600 mM KCl; lane 13 = pooled fractions eluted between 600 and 900 mM KCl. IRP1 = iron-regulatory protein 1.
Purified IRP1 is biologically active. A, IRP1 activity of fractions eluted from the FPLC was assessed by electrophoretic mobility shift assay. Two micrograms of each purified IRP1 fraction was incubated with 0.1 ng [32P]-IRE RNA probe (15,000 cpm) and subjected to non-denaturing electrophoresis on 6% acrylamide gel. The gel was dried and analyzed with the "PhorphorImager" software. Lanes 1-7 correspond, respectively, to fractions 5-11 from Figure 4. Numbers below the lanes represent the percentage of IRP1 activity for bands without EtSH, compared to those with EtSH (100% activation). B, IRP1 activity is inhibited by the addition of FeSO4 and cysteine. Twenty nanograms purified IRP1 was treated with 50 µM FeSO4 and 10 mM cysteine for 30 min at 22°C, and IRE-binding activity was assayed by electrophoretic mobility shift assay. IRP1 = iron-regulatory protein 1; IRE = iron-responsive element; EtSH = ß-mercaptoethanol.
Acknowledgments
We are thankful to Dr. Sonia Gurgueira (Universidade São Francisco, Bragança Paulista, SP, Brazil) for valuable discussions and suggestions.
Address for correspondence: H. Carvalho, Departamento de Ciências Biológicas, Universidade Federal de São Paulo, Campus Diadema, R. Prof. Artur Ridel, 275, 09972-270 Diadema, SP, Brasil. Fax: +55-11-4043-6428. E-mail: helotonio.carvalho@gmail.com
Research supported by FAPESP and CNPq. H. Carvalho is the recipient of a fellowship from FAPESP. Received July 6, 2007. Accepted December 11, 2007.
- 1. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine 3rd edn. New York: Oxford University Press; 1999.
- 2. Thorstensen K, Romslo I. The role of transferrin in the mechanism of cellular iron uptake. Biochem J 1990; 271: 1-9.
- 3. Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1996; 1275: 161-203.
- 4. Mullner EW, Neupert B, Kuhn LC. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell 1989; 58: 373-382.
- 5. Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5' untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci U S A 1988; 85: 2171-2175.
- 6. Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell 1993; 72: 19-28.
- 7. Hentze MW, Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci U S A 1996; 93: 8175-8182.
- 8. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004; 117: 285-297.
- 9. Klausner RD, Harford JB. cis-trans models for post-transcriptional gene regulation. Science 1989; 246: 870-872.
- 10. Guo B, Yu Y, Leibold EA. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J Biol Chem 1994; 269: 24252-24260.
- 11. Constable A, Quick S, Gray NK, Hentze MW. Modulation of the RNA-binding activity of a regulatory protein by iron in vitro: switching between enzymatic and genetic function? Proc Natl Acad Sci U S A 1992; 89: 4554-4558.
- 12. Martins EA, Robalinho RL, Meneghini R. Oxidative stress induces activation of a cytosolic protein responsible for control of iron uptake. Arch Biochem Biophys 1995; 316: 128-134.
- 13. Pantopoulos K, Hentze MW. Rapid responses to oxidative stress mediated by iron-regulatory protein. EMBO J 1995; 14: 2917-2924.
- 14. Carvalho H, Bechara EJ, Meneghini R, Demasi M. Haem precursor delta-aminolaevulinic acid induces activation of the cytosolic iron-regulatory protein 1. Biochem J 1997; 328 (Part 3): 827-832.
- 15. Pantopoulos K, Hentze MW. Nitric oxide signaling to iron-regulatory protein: direct control of ferritin mRNA translation and transferrin receptor mRNA stability in transfected fibroblasts. Proc Natl Acad Sci U S A 1995; 92: 1267-1271.
- 16. Castro LA, Robalinho RL, Cayota A, Meneghini R, Radi R. Nitric oxide and peroxynitrite-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblasts. Arch Biochem Biophys 1998; 359: 215-224.
- 17. Eisenstein RS, Tuazon PT, Schalinske KL, Anderson SA, Traugh JA. Iron-responsive element-binding protein. Phosphorylation by protein kinase C. J Biol Chem 1993; 268: 27363-27370.
- 18. Lin K, Kurland I, Xu LZ, Lange AJ, Pilkis J, el-Maghrabi MR, et al. Expression of mammalian liver glycolytic/gluconeogenic enzymes in Escherichia coli: recovery of active enzyme is strain and temperature dependent. Protein Expr Purif 1990; 1: 169-176.
- 19. Weickert MJ, Doherty DH, Best EA, Olins PO. Optimization of heterologous protein production in Escherichia coli Curr Opin Biotechnol 1996; 7: 494-499.
- 20. Yasukawa T, Kanei-Ishii C, Maekawa T, Fujimoto J, Yamamoto T, Ishii S. Increase of solubility of foreign proteins in Escherichia coli by coproduction of the bacterial thioredoxin. J Biol Chem 1995; 270: 25328-25331.
- 21. Amrein KE, Takacs B, Stieger M, Molnos J, Flint NA, Burn P. Purification and characterization of recombinant human p50csk protein-tyrosine kinase from an Escherichia coli expression system overproducing the bacterial chaperones GroES and GroEL. Proc Natl Acad Sci U S A 1995; 92: 1048-1052.
- 22. Sareen D, Sharma R, Vohra RM. Chaperone-assisted overexpression of an active D-carbamoylase from Agrobacterium tumefaciens AM 10. Protein Expr Purif 2001; 23: 374-379.
- 23. Goenka S, Rao CM. Expression of recombinant zeta-crystallin in Escherichia coli with the help of GroEL/ES and its purification. Protein Expr Purif 2001; 21: 260-267.
- 24. Mitsuda M, Iwasaki M. Improvement in the expression of CYP2B6 by co-expression with molecular chaperones GroES/EL in Escherichia coli Protein Expr Purif 2006; 46: 401-405.
- 25. Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 2002; 295: 1852-1858.
- 26. Keskin O, Bahar I, Flatow D, Covell DG, Jernigan RL. Molecular mechanisms of chaperonin GroEL-GroES function. Biochemistry 2002; 41: 491-501.
- 27. Ewalt KL, Hendrick JP, Houry WA, Hartl FU. In vivo observation of polypeptide flux through the bacterial chaperonin system. Cell 1997; 90: 491-500.
- 28. Gray NK, Quick S, Goossen B, Constable A, Hirling H, Kuhn LC, et al. Recombinant iron-regulatory factor functions as an iron-responsive-element-binding protein, a translational repressor and an aconitase. A functional assay for translational repression and direct demonstration of the iron switch. Eur J Biochem 1993; 218: 657-667.
- 29. Bradford MA. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248-254.
- 30. Hentze MW, Rouault TA, Harford JB, Klausner RD. Oxidation-reduction and the molecular mechanism of a regulatory RNA-protein interaction. Science 1989; 244: 357-359.
- 31. Konarska MM, Sharp PA. Electrophoretic separation of complexes involved in the splicing of precursors to mRNAs. Cell 1986; 46: 845-855.
- 32. Brazzolotto X, Gaillard J, Pantopoulos K, Hentze MW, Moulis JM. Human cytoplasmic aconitase (iron regulatory protein 1) is converted into its [3Fe-4S] form by hydrogen peroxide in vitro but is not activated for iron-responsive element binding. J Biol Chem 1999; 274: 21625-21630.
- 33. Dupuy J, Volbeda A, Carpentier P, Darnault C, Moulis JM, Fontecilla-Camps JC. Crystal structure of human iron-regulatory protein 1 as cytosolic aconitase. Structure 2006; 14: 129-139.
- 34. Walden WE, Selezneva AI, Dupuy J, Volbeda A, Fontecilla-Camps JC, Theil EC, et al. Structure of dual function iron-regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006; 314: 1903-1908.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
14 Feb 2008 -
Date of issue
Apr 2008
History
-
Received
06 July 2007 -
Accepted
11 Dec 2007