Abstract
Oxysterols are 27-carbon atom molecules resulting from autoxidation or enzymatic oxidation of cholesterol. They are present in numerous foodstuffs and have been demonstrated to be present at increased levels in the plasma of patients with cardiovascular diseases and in atherosclerotic lesions. Thus, their role in lipid disorders is widely suspected, and they might also be involved in important degenerative diseases such as Alzheimer's disease, osteoporosis, and age-related macular degeneration. Since atherosclerosis is associated with the presence of apoptotic cells and with oxidative and inflammatory processes, the ability of some oxysterols, especially 7-ketocholesterol and 7β-hydroxycholesterol, to trigger cell death, activate inflammation, and modulate lipid homeostasis is being extensively studied, especially in vitro. Thus, since there are a number of essential considerations regarding the physiological/pathophysiological functions and activities of the different oxysterols, it is important to determine their biological activities and identify their signaling pathways, when they are used either alone or as mixtures. Oxysterols may have cytotoxic, oxidative, and/or inflammatory effects, or none whatsoever. Moreover, a substantial accumulation of polar lipids in cytoplasmic multilamellar structures has been observed with cytotoxic oxysterols, suggesting that cytotoxic oxysterols are potent inducers of phospholipidosis. This basic knowledge about oxysterols contributes to a better understanding of the associated pathologies and may lead to new treatments and new drugs. Since oxysterols have a number of biological activities, and as oxysterol-induced cell death is assumed to take part in degenerative pathologies, the present review will focus on the cytotoxic activities of these compounds, the corresponding cell death signaling pathways, and associated events (oxidation, inflammation, and phospholipidosis).
Apoptosis; Atherosclerosis; Inflammation; Oxysterols; Phospholipidosis
Braz J Med Biol Res, July 2008, Volume 41(7) 545-556 (Review)
Side effects of oxysterols: cytotoxicity, oxidation, inflammation, and phospholipidosis
A. Vejux1, L. Malvitte2,3 and  Correspondence and Footnotes
Correspondence and Footnotes
 G. Lizard3
G. Lizard3
1Unité Transporter in Imaging and Radiotherapy in Oncology (TIRO), Commissariat à l'Energie Atomique, DSV-DIEP-SBTN, Faculté de Médicine, Université de Nice Sophia Antipolis, Nice, France
2Service d'Ophtalmologie, Centre Hospitalier Universitaire de Dijon, Hôpital Général, Dijon, France
3Centre de Recherche Inserm U866, Université de Bourgogne, Faculté des Sciences Gabriel, Dijon, France
 Text
Text
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Oxysterols are 27-carbon atom molecules resulting from autoxidation or enzymatic oxidation of cholesterol. They are present in numerous foodstuffs and have been demonstrated to be present at increased levels in the plasma of patients with cardiovascular diseases and in atherosclerotic lesions. Thus, their role in lipid disorders is widely suspected, and they might also be involved in important degenerative diseases such as Alzheimer's disease, osteoporosis, and age-related macular degeneration. Since atherosclerosis is associated with the presence of apoptotic cells and with oxidative and inflammatory processes, the ability of some oxysterols, especially 7-ketocholesterol and 7β-hydroxycholesterol, to trigger cell death, activate inflammation, and modulate lipid homeostasis is being extensively studied, especially in vitro. Thus, since there are a number of essential considerations regarding the physiological/pathophysiological functions and activities of the different oxysterols, it is important to determine their biological activities and identify their signaling pathways, when they are used either alone or as mixtures. Oxysterols may have cytotoxic, oxidative, and/or inflammatory effects, or none whatsoever. Moreover, a substantial accumulation of polar lipids in cytoplasmic multilamellar structures has been observed with cytotoxic oxysterols, suggesting that cytotoxic oxysterols are potent inducers of phospholipidosis. This basic knowledge about oxysterols contributes to a better understanding of the associated pathologies and may lead to new treatments and new drugs. Since oxysterols have a number of biological activities, and as oxysterol-induced cell death is assumed to take part in degenerative pathologies, the present review will focus on the cytotoxic activities of these compounds, the corresponding cell death signaling pathways, and associated events (oxidation, inflammation, and phospholipidosis).
Key words: Apoptosis; Atherosclerosis; Inflammation; Oxysterols; Phospholipidosis
Text

Oxysterols: sources and pathophysiological relevance
Oxysterols are 27-carbon atom cholesterol oxidation products resulting from either the autoxidation or enzymatic oxidation of oxysterols (1,2). Cholesterol is composed of three regions: a hydrocarbon tail (also called the lateral chain), a ring structure region with four hydrocarbon rings (A, B, C, and D), and a hydroxyl group. Oxidation can occur on the ring structure or on the lateral chain. The double bond present on the B hydrocarbon ring can be the target of free radical attacks, and therefore positions 4, 5, 6 and 7 are the most sensitive sites for autoxidation. The autoxidation of cholesterol on the B hydrocarbon ring leads to the formation of 7α- and 7β-hydroxycholesterol, 7-ketocholesterol (constituting a good autoxidation marker), 5α,6α-epoxycholesterol, 5β,6β-epoxycholesterol, and cholestane-3β,5α, 6β-triol (Figure 1). It is noteworthy that introducing an oxygen function increases the rate of cholesterol degradation into more polar compounds, and oxysterols with additional oxygen functions on the lateral chain can therefore be easily transported out of cells and eliminated.
Oxysterols are present in various foodstuffs, notably cholesterol-rich foods such as dairy products, milk, eggs, dried egg powder, clarified butter (or ghee), meat products, and dried or stored fish (1). Generally, products containing cholesterol are susceptible to oxidation, especially dehydrated foodstuffs subjected to radiation or submitted to high temperatures, as well as those that are cooked in the presence of oxygen (1). Indeed, under these conditions, alimentary cholesterol is exposed to numerous reactive oxygen species (ROS), such as singlet oxygen (1O2), which can contribute to the generation of cholesterol hydroperoxides (3), hydrogen peroxide (H2O2), hydroxyl radical (·OH), and ozone (O3). Consequently, when food is not stored for lengthy periods in an environment other than in a vacuum, the generation of oxysterols markedly increases. The most commonly detected oxysterols in processed foods are 7-oxygenated sterols (7-ketocholesterol, 7α-hydroxycholesterol, 7β-hydroxycholesterol), 5,6-oxygenated sterols (5α, 6α-epoxycholesterol, 5β,6β-epoxycholesterol, cholestane-3β,5α, 6β-triol), as well as 25-hydroxycholesterol, 19-hydroxycholesterol, 20α-hydroxycholesterol, 3β-hydroxy-5α-cholestane-6-one, and 3β, 5α-dihydroxycholestane-6-one, which are present in smaller amounts (1).
Food oxysterols are mainly absorbed as esters in the intestinal tract and further transported in the plasma by chylomicrons. Interestingly, contrary to 7β-hydroxycholesterol, the 7-ketocholesterol is slightly absorbed and rapidly metabolized by the liver (4). Furthermore, in the plasma, oxysterols are transported by lipoproteins. The highest levels of oxysterols are present in low-density lipoproteins (LDL) and to a lesser extent in high-density lipoproteins and very-low-density lipoproteins. Some studies have also shown that oxysterols can be transported by albumin (1). Compared to cholesterol, oxysterols are absorbed more quickly in the intestine, have faster plasma clearance and are quickly collected by tissues. It should be noted that cholesterol autoxidation can also take place in various tissues, especially in the arterial wall, during the atheromatous process (1,2,5).
The ability of tissues to promote oxysterol synthesis was reported for the first time in 1956 (1). To this end, radiolabeled cholesterol was incubated with a subcellular fraction enriched in mitochondria, and 25-hydrocholesterol and 26-hydrocholesterol were formed. The enzymatic synthesis of another oxysterol, 24-hydroxycholesterol, was described much later in 1974 (1). Since even minor oxidation of cholesterol during sample processing would yield a substantial increase in oxysterol levels, isotopic methods combined with gas chromatography and mass spectrometry were developed to avoid artefactual results (1). Consequently, it is now accepted that among the oxysterols found in plasma, some of them, such as 7a-hydroxycholesterol, 24S-hydroxycholesterol, 25-hydroxycholesterol, and 27-hydroxycholesterol, can result from enzymatic synthesis.
The microsomal cytochrome P450 system is largely responsible for the enzymatic generation of endogenous oxysterols (Table 1), including hepatic 7α-hydroxycholesterol (catalyzed by CYP7A1), 24S-hydroxycholesterol in brain and retina (catalyzed by CYP46) (6), and 27-hydroxycholesterol in most tissues (catalyzed by CYP27), whereas the minor oxysterol, 25-hydroxycholesterol, is generated by the non-heme iron protein enzyme, cholesterol 25-hydroxylase (7). In the hepatocytes, 27-hydroxylase (CYP27A1) and 7α-hydroxylase (CYP7A1) are two key enzymes of bile acid biosynthesis, and the enzyme CYP7A1 is the enzyme limiting the conversion of cholesterol into bile acids (1,2,7). The enzyme CYP27A1 also catalyses the addition of a hydroxyl group not only on cholesterol to give 27-hydroxycholesterol, but also on a number of intermediates in bile synthesis, such as the 5β-cholestane-3α, 7α, 12α-triol and the 5β-cholestane-3α, 7α-diol, to produce their hydroxylated forms in position 27. In treatment with phenobarbital, a hepatic production of 4β-hydroxycholesterol resulting from the activation of the enzyme CYP3A4 has also been described (1). Physiologically, distinct strategies exist to reverse important accumulation of oxidized sterols. Thus, 7-ketocholesterol is the substrate for CYP27A1, which is expressed in the liver and a range of extrahepatic tissues, including macrophages (1,2). 7β-hydroxycholesterol can be metabolized by the enzyme 11β-hydroxysteroid dehydrogenase type 1 (4). In hamster and chicken microsome preparations, an enzymatic conversion of 7α-hydroxycholesterol into 7-ketocholesterol has been reported (8). It has also been found that β-amyloid, the toxic peptide in neurons of Alzheimer's disease patients, binds oxysterols and catalyzes its oxidation to 7β-hydroxycholesterol (9). In atherosclerotic arteries, evidence for O3 formation leading subsequently to autoxidation of cholesterol and to oxysterols formation has also been reported (10), but remains questionable (11). Thus, whereas oxysterols produced by cholesterol ozonolysis have been described, and have biological activities (12), the part taken by O3, and by these molecules in atherosclerosis and other degenerative diseases requires further investigation.
It is well established that oxysterols are involved in some physiological processes, such as the regulation of cholesterol homeostasis (13). However, these compounds are also suspected to play key roles in various pathologies, mostly in cardiovascular diseases (5). Indeed, 7-ketocholesterol and 7β-hydroxycholesterol, which are major components of oxidized LDL (oxLDL), are known to contribute to the genesis of atherosclerosis (14), and are found at increased levels in atherosclerotic lesions, and in the plasma of patients with cardiovascular diseases and of subjects after a fat-rich meal (1,5,14). A number of investigations have also demonstrated that oxysterols can have cytotoxic and pro-inflammatory activities (15), and some studies have shown that these molecules can stimulate the differentiation of mesenchymal cells, monocytes, keratinocytes, lens epithelial cells, and osteoblasts (16). Therefore, in addition to atherosclerosis, it is now suggested that oxysterols might contribute to the development of numerous other degenerative diseases such as age-related macular degeneration (17), Alzheimer's disease (18), and osteoporosis (19).
Oxysterol-induced cell death and associated signaling pathways
Cell death is a fundamental physiological process that plays important roles in ontogenesis and in tissue homeostasis. It is now accepted that its dysregulation can contribute to the emergence of numerous diseases. From 1965, two distinct types of cell death have been described: apoptosis (also called programmed cell death) and necrosis (also called oncosis). Apoptosis is induced by many biological, chemical and physical agents, and it is involved in embryonic development. It is morphologically defined by a decrease in cell volume, maintenance of membrane integrity, and condensation and/or fragmentation of nuclei. Two main signaling pathways capable of triggering apoptosis have been described: 1) the death receptor pathway, which belongs to the TNF superfamily pathway, leads to a direct activation of caspases (cysteinyl-aspartate-cleaving proteases) existing as pro-enzymes or zymogen, and 2) the mitochondrial signaling pathway. The mitochondrial pathway involves the formation of the apoptosome, subsequent to mitochondrial depolarization, which is considered to be a point of no return, and the activation of a cascade of caspases contributing to numerous morphological and biochemical modifications, including internucleosomal DNA fragmentation. In certain cases, secondary necrosis is the final step of apoptosis. Necrosis is characterized by swelling of cells and organites and increased cell permeability (20).
Currently, several other types of cell death have been described (21): apo-necrosis, autophagy, pyroptosis, and paraptosis. Among these additional forms of cell death, autophagy has been extensively studied (22). It contributes to the renewal of proteins, RNAs, and other cytoplasmic macromolecules. During autophagy, the cytoplasmic components are sequestered into cytoplasmic structures with a double membrane 300-900 nm in diameter (23). Autophagy can be induced under various stress conditions: amino acid deprivation and treatment with rapamycin or steroid hormones.
Since some dead cells, including apoptotic cells, are present in atherosclerotic lesions (1,5), the type of cell death induced by oxysterols present at increased levels in atherosclerotic plaque (especially 7-ketocholesterol and 7β-hydroxycholesterol) is widely studied in vascular wall cells.
Whereas most investigations report that oxysterols are potent inducers of apoptosis, they can also induce necrosis in various cell types (24,25), and this mode of cell death may be more pronounced at higher oxysterol concentrations. Generally, 7-ketocholesterol, 7β-hydroxycholesterol, and 27-hydroxycholesterol induce apoptosis on various cell types, whereas 25-hydroxycholesterol has a more or less pronounced cytotoxicity depending on the cell considered (15,26). Moreover, the ability of 7-ketocholesterol to induce an apoptotic mode of cell death associated with autophagic characteristics in U937 cells and in human aortic smooth muscle cells has been reported based on increased fluorescence staining with monodansylcadaverine in dying cells (27) and on the analysis of the two forms of the LC3 protein (microtubule-associated protein 1 light chain 3) (28), respectively. LC3-I is cytosolic, whereas LC3-II, identified in 7-ketocholesterol-treated cells (28), is linked to the plasma membrane and found in autophagic vacuoles. Mutation analyses suggest that LC3-I is formed by the removal of the C-terminal 22 amino acids from newly synthesized LC3, followed by the conversion of a fraction of LC3-I into LC3-II (23). The amount of LC3-II is correlated with the extent of autophagosome formation (23). Whereas the involvement of an autophagic process is supported by the identification of LC3-II in 7-ketocholesterol-treated human aortic smooth muscle cells, the presence of cytoplasmic structures stained by the lysosome cationic lipophilic dye, monodansylcadaverine, which is detected before the loss of mitochondrial potential in 7-ketocholesterol-treated U937 cells, suggests instead that 7-ketocholesterol is a potent inducer of phospholipidosis (29). Indeed, under treatment with cytotoxic oxysterols, these structures, which are identified both in cells with condensed and/or fragmented nuclei characteristic of apoptotic cells and in cells with swollen nuclei considered as oncotic cells, can also be stained with Nile Red, which colors neutral and polar lipids yellow and orange/red, respectively (30). In addition, their biochemical characterization revealed that they contain a high concentration of phosphatidylcholine and sphingomyelin. Moreover, at cytoplasmic levels, since oxysterol-induced cell death is associated with important modifications (presence of multilamellar cytoplasmic structures) revealed by transmission electron microscopy (27,30,31), we asked whether 7-ketocholesterol-induced apoptosis was associated with an activation of reticulum, stress inducing a coordinated activated program called the unfolded protein response (UPR). In 7-ketocholesterol-treated human aortic smooth muscle cells, an activation of the UPR pathway characterized by the following events was observed (32): early transient calcium oscillations and induction of the expression of the cell death effector CHOP and GRP78/Bip chaperone via the activation of IRE-1, all hallmarks of the UPR. It was also shown that 7-ketocholesterol activated the IRE-1/Jun-NH2-terminal kinase/AP-1 signaling pathway to promote expression of Nox-4, an ROS, generating NAD(P)H oxidase homologue.
Currently, whereas the pre-mitochondrial events associated with oxysterol-induced cell death are still not well known, early modifications of the cytoplasmic membrane, calcium influx, and overproduction of oxygen radicals seem to play important roles. Indeed, some pro-apoptotic oxysterols have the ability to modify biophysical membrane properties and the composition of raft domains involved in signal transduction (33); some of them also induce intracellular calcium oscillations that can be blocked by calcium channel blockers such as verapamil and nifedipine (34). The role of calcium influx in 7 ketocholesterol-induced PC12 cell death was also supported by the activation of calmodulin, a ubiquitous calcium-binding protein that can bind up to four calcium ions, which contributes to the loss of mitochondrial transmembrane potential, the release of cytochrome c, and subsequent activation of caspase-3 (34). In addition, in 25-hydroxycholesterol-treated fibroblasts and macrophages, calcium influx is associated with activation of the cytosolic phospholipase-A2 and with arachidonic acid release. This activation could be, at least in part, capable of triggering apoptosis, since in PLA2 (-/-) cells, the cytotoxic effect of 25-hydroxycholestrol is substantially reduced (35).
Interestingly, among the signaling pathways capable of triggering oxysterol-induced apoptosis, some investigations report the involvement of the mitochondrial pathway and the probable participation of the death receptor pathway. Thus, in 25-hydroxycholesterol- and 7β-hydroxycholesterol-treated cells or in treatment with oxLDL, increased levels of Fas and Fas ligand and their corresponding mRNAs have been reported; these latter might contribute to activating apoptosis (36). Consequently, given that the death of smooth muscle cells is a key element in the destabilization of the fibrous cap, the simultaneous presence of 7-ketocholesterol and of Fas ligand could increase the risk of rupture of atherosclerotic plaque (37). In the presence of 7-ketocholesterol, a predisposition to the Fas and to the TNF-α signaling pathway has also been described in human aortic smooth muscle cells (37), and since 7-ketocholesterol and TNF-α have been identified in atherosclerotic lesions, their simultaneous presence might also contribute to plaque destabilization.
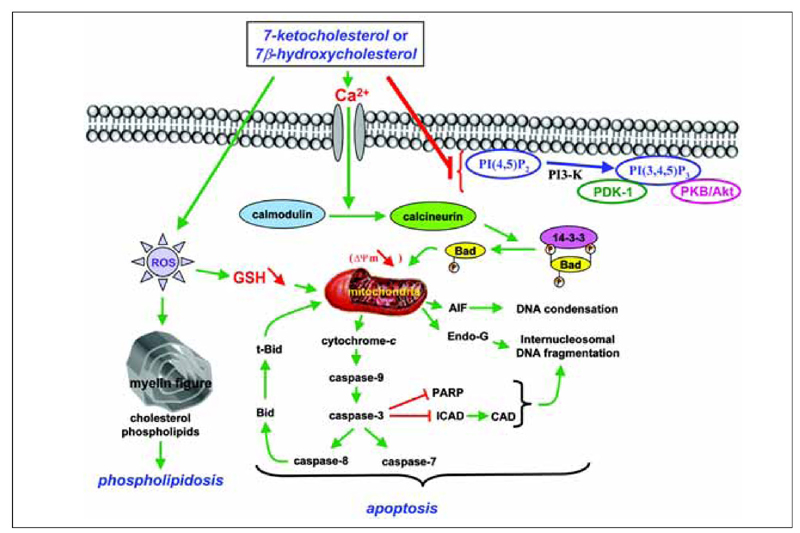
In treatment of U937 and THP-1 cells and vascular cells (endothelial cells, smooth muscle cells) with 7-ketocholesterol and 7β-hydroxycholesterol, some investigations demonstrated that the mitochondrial pathway was involved. This pathway is characterized by a loss of transmembrane mitochondrial potential (ΔΨm), a mitochondrial release of various proteins (cytochrome c, apoptosis-inducing factor, and endonuclease G) into the cytosol, negative regulation of Bcl-2 expression, Bad dephosphorylation, Bid cleavage, activation of different caspases (caspase-2, -3, -7, -8, and -9), inhibitor caspase-activated deoxyribonuclease cleavage, poly-ADP-ribose polymerase degradation, internucleosomal DNA fragmentation, and condensation and/or fragmentation of the nuclei (38,39). During treatment with 7β-hydroxycholesterol and 25-hydroxycholesterol, an inactivation of the Akt/PKB pathway was observed in U937 cells and in murine macrophages, respectively (40). Given that down-regulation of the PI3-K/PDK-1/Akt/PKB signaling pathway is a vitamin E inhibitable event associated with 7-ketocholesterol-induced apoptosis, these data suggest relationships between mitochondria and the Akt/PKB signaling pathway since mitochondrial depolarization can be inhibited by vitamin E (41). A proposed model of the different signaling pathways induced by 7-ketocholesterol, which is probably the most extensively studied oxysterol, is presented in Figure 2.
When oxysterol mixtures, prepared according to the relative proportions and molar concentrations found in human atheroma lesions, were used, contradictory results were obtained. Therefore, although an oxysterol mixture composed of 7α-hydroxycholesterol (5%), 7β-hydroxycholesterol (10%), cholesterol-5α, 6α-epoxide (20%), cholesterol-5β,6β-epoxide (20%), cholestane-3β,5α, 6β-triol (9%), 7-ketocholesterol (35%), and 25-hydroxycholesterol (1%) had no cytotoxic effects (42), the combination of four oxysterols [used in atheroma relative proportions: 27-hydroxycholesterol (23.0), 7-ketocholesterol (16.8), 7β-hydroxycholesterol (9.52), and 25-hydroxycholesterol (1.0)] was pro-apoptotic (43). These observations, obtained under in vitro experimental conditions more precisely mimicking the physiopathological environments observed in atherosclerotic lesions, not only underscore the potent side effects of oxysterols, but also focus on the complexity of the resulting cytotoxic characteristics of an oxysterol mixture, which depends on the oxysterols considered, their levels, and their relative proportions.
Oxysterols: potent pro-oxidative molecules
Epidemiological investigations and animal experiments have provided evidence supporting the role of lipid peroxidation in atherogenesis and cardiovascular diseases, age-related macular degeneration, and Alzheimer's disease (5,9,17). Some studies have indicated that oxLDL, known to contain high levels of oxysterol, and some oxysterols identified in atherosclerotic lesions or entering retinal pigmentary epithelial cells through choriocapillaris (5,14,17), are relevant in the induction of oxidative processes resulting from an overproduction of ROS. ROS are either free radicals such as superoxide anion (O2·_) and hydroxyl radical (·OH), reactive anions containing oxygen atoms such as H2O2 and peroxynitrite (ONOOH), and/or secondary radicals [alkyl (R·), alkoxyl (RO·) and peroxyl (ROO·)], which are chemically activated by ROS following the oxidizing activity of ROS on unsaturated fatty acids, carbohydrates, and/or proteins; they can, in turn, contribute to producing free radicals. It is worth noting that these ROS have important side effects on vascular wall cells at various stages of the development of atherosclerosis as well as on retinal and neuronal cells. Indeed, they favor certain major pro-atherosclerotic events: proliferation of smooth muscle cells; enhanced accumulation of oxLDL in macrophages and smooth muscle cells as well as subsequent formation of foam cells; loss of endothelial cell reactivity; peroxidation of membrane lipids and accumulation of lipid oxidation products such as malondialdehyde and oxysterols resulting from the autoxidation of cellular cholesterol (1); induction of cell death (5).
Therefore, to evaluate the oxidative potential of oxLDL and oxysterols, which are strongly suspected of being involved in various pathological processes, some investigations have been conducted in vivo and especially in vitro with oxysterols and oxysterol mixtures. Rats fed a diet containing oxysterols have significantly higher mRNA levels of glutathione peroxidase and superoxide dismutase, significantly enhanced glutathione peroxidase activity, and lower concentrations of total and reduced glutathione in the liver than rats fed without oxysterols, suggesting that dietary oxysterols stress the antioxidant defense system in rats (44). In atherosclerotic apolipoprotein E-deficient mice, it has also been reported that some oxysterols (7-ketocholesterol, 7β-hydroxycholesterol, and cholesterol-5β,6β-epoxide) induced the activation of macrophage NADPH-oxidase, arachidonic acid release, and superoxide anion production, which contribute to enhancing cell-mediated oxidation of LDL, and that vitamin E was able to counteract these different events (45). In vitro, under treatment with 7-ketocholesterol, 7β-hydroxycholesterol, cholesterol-5β,6β-epoxide, cholesterol-5α, 6α-epoxide, and 25-hydroxycholesterol, important oxidative processes, sometimes associated with a complex mode of cell death with some of the characteristics of apoptosis, have also been described on various cell types (15,27,32). Thus, on 7-ketocholesterol-treated U937 cells, an abrupt drop in reduced glutathione associated with an increased production of ROS preceding caspase activation has been reported (46). In human aortic smooth muscle cells, 7-ketocholesterol also induces oxidative stress associated with apoptotic events, and it has been demonstrated that this specific effect of 7-ketocholesterol is mediated by a robust upregulation (3-fold from the basal level) of Nox-4, an ROS-generating NAD(P)H oxidase homologue (32).
Given that cytotoxic concentrations of 7-ketocholesterol and 7β-hydroxycholesterol also induce lipid peroxidation processes and down-regulation of anti-oxidative defenses (46), genotoxic effects of these compounds have been observed (47). Interestingly, some antioxidants [apigenin, astaxanthin, carotene, folate, glutathione, lycopene, N-acetylcysteine, vitamin E (α-tocopherol)] have been described as preventing oxysterol-induced cell death (27,41,46). Oxysterol mixtures, in atheroma-relevant proportions, also display pro-oxidative activities. Thus, when 7β-hydroxycholesterol is combined with cholesterol-5β,6β-epoxide, the decrease in the glutathione level in U937 cells is significantly greater than with 7β-hydroxycholesterol alone (48). Subsequent experiments showed that when 7-ketocholesterol was administered to cells together with another oxysterol, namely 7β-hydroxycholesterol, the generation of ROS in J774-A1 murine macrophages was markedly attenuated (42).
It has been proposed that competition among oxysterols, apparently at the level of NADPH oxidase, diminishes the ROS induction and direct toxicity that is evoked by specific oxysterols. Moreover, in the J774-A1 murine macrophage cell line, with a biologically representative oxysterol mixture (5-cholestene-3β,7α-diol (10.2%), 5-cholestene-3β,7β-diol (6.7%), cholestane-5α,6α-epoxy-3β-ol (10.3%), cholestane-5β,6β-epoxy-3β-ol (24.3%), cholestane-3β, 5α, 6β-triol (10.0%), 5-cholestene-3β-ol-7-one (31.0%), and 5-cholestene-3β,25diol (7.3%)), which approximates the proportions of oxysterols found in LDL from human plasma, the mRNA level of manganese superoxide dismutase was markedly increased after 24-h exposure. This overexpression of manganese superoxide dismutase mRNA may serve as an important adaptive response of oxysterols to regulate oxygen species involved in the apoptotic death of macrophage/foam cells. However, in certain conditions, anti-oxidative effects of oxysterols have also been described. Thus, oxidized cholesterol in oxLDL may be responsible for the inhibition of lipopolysaccharide (LPS)-induced nitric oxide production in J774-A1 murine macrophages (49).
It has also been reported that a specific oxysterol combination consisting of 22(S)- or 22(R)-hydroxycholesterol and 20(S)-hydroxycholesterol has potent osteogenic properties in vitro when applied to osteoprogenitor cells, including M2-10B4 marrow stromal cells, and that this osteogenic combination of oxysterols prevents adverse effects of oxidative stress on the differentiation of M2 cells into mature osteoblastic cells (50). Under these conditions, the protective effect of the oxysterols against oxidative stress was dependent on cyclooxygenase 1 and was associated with the osteogenic property of the oxysterols (50).
Oxysterols and inflammatory processes
It is generally accepted that vascular areas of atherosclerotic progression are in a state of persistent inflammation, and the association of hypercholesterolemia with atherosclerosis is well established. However, the primary means by which a long-term excess of blood cholesterol contributes to the expression of inflammatory events characterizing the onset and/or progression of atherosclerosis is still largely undefined. Given that numerous oxidative events are associated with the development of atherosclerotic plaque, it is now accepted that oxLDL plays a major role in the initiation and promotion of fatty streaks and fibrotic plaques.
Among oxidative agents present in oxLDL that may be responsible for inflammatory processes, some oxysterols appear to be particularly attractive targets for research. When human monocytes, monocyte-derived macrophages (THP-1 cells), and porcine retinal pigment epithelial cells were exposed to a series of different oxysterols (7-ketocholesterol, 7β-hydroxycholesterol, 24-hydroxycholesterol, 25-hydroxycholesterol, or cholestane-3β,5α, 6β-triol), all of them had a tendency to stimulate IL-8 production, but 25-hydroxycholesterol was the most potent (15). It is worth noting that in human mononuclear cells isolated by Ficoll-Paque centrifugation, the pro-inflammatory activity of 25-hydroxycholesterol was enhanced in hypoxic conditions, and this oxysterol was also capable of potentiating LPS-induced IL-1β secretion (51). Moreover, in human promonocytic leukemia cells (U937) and the J774-A1 murine macrophage cell line, a biologically representative oxysterol mixture (7α-hydroxycholesterol (5%), 7β-hydroxycholesterol (10%), cholesterol a-epoxide (20%), cholesterol β-epoxide (20%), cholestane-3β,5α, 6β-triol (9%), 7-ketocholesterol (35%), and 25-hydroxycholesterol (1%)) induced both expression and synthesis of key chemokines for monocytes/macrophages, especially MCP-1 (52), and upregulated TGF-β1 expression, playing a pivotal role in smooth muscle cell chemoattraction and differentiation into myofibroblast-like structures (53). In U937 and THP-1 cells, stimulation of the secretion of various chemokines involved in the recruitment of immunocompetent cells at the subendothelial level, namely MCP-1, MIP-1β, TNF-α, IL-1β and IL-8, was also reported, mainly in the presence of 7β-hydroxycholesterol and 25-hydroxycholesterol (54). Treatment of aortic rat smooth muscle cells with 25-hydroxycholesterol or 22R-hydroxycholesterol causes the accumulation of group IIA secretory phospholipase A(2) and increases enzyme activity (55). However, under certain conditions, anti-inflammatory effects of oxysterols have also been reported. Thus, when human macrophages were incubated with 7-ketocholesterol, 7β-hydroxycholesterol, 25 hydroxycholesterol, or 27-hydroxycholesterol before exposure to LPS, all oxysterols, except 7-ketocholesterol, significantly decreased LPS-induced TNF-α secretion (56). Currently, little is known about the molecular mechanisms involved in oxysterol-induced inflammation at the transcriptional level. Various modes of regulation of gene expression are probably involved since the activation of LXR receptors can only be envisaged with a few pro-inflammatory oxysterols, namely with 22R-hydroxycholesterol and 25-hydroxycholesterol, but not with 7β-hydroxycholesterol, which is not an LXR substrate. Similarly, little information is available on oxysterol-activated metabolic pathways at the translational and post-translational levels. However, in 7β-hydroxycholesterol-treated U937 and THP-1 cells, it has been clearly established that IL-8 secretion is associated with activation of the MEK/ERK1/2 signaling pathway (54). Interestingly, in the presence of PD98059, a potent inhibitor of MEK, IL-8 secretion was totally counteracted, whereas no decrease in the IL-8 mRNA level or intracellular IL-8 was observed (54).
Cytotoxic oxysterols: powerful inducers of phospholipidosis
As atherosclerosis is characterized at the subendothelial level by an intracellular accumulation of lipids in macrophages and smooth muscle cells, and since these cells can transform into foam cells _ both lipid-laden cells characteristic of the first step of atherosclerosis _ it is important to clarify the function of certain oxysterols in intracellular lipid accumulation, especially with oxysterols such as 7-ketocholesterol and 7β-hydroxycholesterol present at increased levels in atherosclerotic plaque and capable of inducing cytotoxic effects (14,15). Indeed, death of macrophage foam cells leads to the development of the lipid core of advanced atherosclerotic lesions, while death of smooth muscle cells thins the fibrous cap, and these changes destabilize lesions, predisposing them to rupture, with thrombogenic consequences. Moreover, it has been reported that 7-ketocholesterol can favor in vitro monocyte differentiation and foam cell formation (16). Interestingly, in the atherosclerotic process, the part played by unesterified cholesterol in intracellular lipid accumulation is well documented, and the associated cellular modifications, such as the presence of multilamellar cytoplasmic structures, recall the modifications observed under treatment with cytotoxic oxysterols (7-ketocholesterol, 7β-hydroxycholesterol, cholesterol-5β, 6β epoxide) in various cell types (24,31, 32,46): presence of multilamellar cytoplasmic structures (also called myelin figures) of various sizes and shapes (27) and intracellular accumulation of polar lipids revealed by Nile Red staining and subcellular fractionation associated with gaseous chromatography coupled with mass spectrometry (30,38). When the cells were treated with 7-ketocholesterol, these biochemical analyses also revealed an accumulation of cholesterol and 7-ketocholesterol in myelin figures. Moreover, with 7-ketocholesterol, it has been reported that myelin figures were localized in acidic compartments revealed by staining with monodansylcadaverine, which is a lysosomotropic cationic fluorescent probe (27). Moreover, when 7-ketocholesterol-treated cells were cultured in the presence of 7-ketocholesterol for periods of time preceding the loss of transmembrane mitochondrial potential considered as a point of no return and reintroduced in 7-ketocholesterol-free medium, the percentage of cells with depolarized mitochondria was substantially reduced and lower levels of polar lipids per cell (evaluated by Nile Red staining and characterized by large red cytoplasmic structures) were found (30).
Taken together, these data demonstrate that 7-ketocholesterol-induced myelin figure formation is associated with the following cellular events: 1) accumulation of polar lipids shown by Nile Red staining and biochemical analysis (high levels of phospholipids, mainly phosphatidylcholine and sphingomyelin in myelin figures isolated after staining with monodansylcadaverine or Nile Red); 2) location of myelin figures in acidic compartments (staining with monodansylcadaverine); 3) accumulation of 7-ketocholesterol in myelin figures; 4) reversibility of polar lipid accumulation. Taken together, these different observations lead us to conclude that 7-ketocholesterol is a potent inducer of phospholipidosis characterized by the following criteria: excessive accumulation of phospholipids in cells; ultrastructural appearance of multilamellar cytoplasmic inclusions, predominantly lysosomal in origin; accumulation of the inducing drug in association with phospholipids in multilamellar structures; reversibility of alterations after interruption of drug treatment (29).
Until now, phospholipidosis has never been described with oxysterol treatment, and the main in vivo and in vitro reports concern phospholipidosis induced by drugs with cationic amphiphilic structures such as amiodarone, fluoxetine, imipramine, chlorcyclizine, tamoxifen, and gentamicin, which are characterized by a hydrophobic ring structure and a hydrophilic side chain with a charged cationic amine group, capable of interacting with lipid cell components and causing lipid storage disorders. Currently, the relationship between phospholipidosis and toxicity remains unclear. It has been suggested that phospholipidosis might be a part of a defense system when the cell is confronted with a substantial accumulation of xenobiotics and their metabolites because the imprisonment of these compounds in the lamellar structures can inactivate their actions (29). Thus, phospholipidosis has been primarily considered as an adaptive response to the exposure of cationic amphiphilic drugs rather than as a toxic response. In addition, multilamellar bodies have been observed to undergo exocytosis and therefore might contribute to reducing accumulation of cytotoxic molecules (29). However, some observations also support the notion that phospholipidosis might be a part of cell death. Indeed, amiodarone and imipramine, which are two major inducers of phospholipidosis, were also shown to inhibit cell proliferation and to activate caspase-mediated apoptosis in cell culture (57). Moreover, in murine macrophages loaded with free cholesterol, some phospholipid whorls, suggesting myelin figures, have been observed, and their presence is associated with considerable cytotoxic effects, including stress of the reticulum connected with the induction of a mode of cell death by apoptosis (58). It has also been suggested, with cytotoxic oxysterols (15) and cationic amphiphilic drugs (29), that the rupture of the acidic compartments containing multilamellar structures might favor the release of some proteolytic enzymes into the cytoplasm, which could subsequently contribute to activating certain apoptotic pathways. Moreover, since vitamin E and caspase inhibitors (z-VAD-fmk: broad-spectrum caspase inhibitor; z-VDVAD-fmk: caspase-2 inhibitor) can counteract phospholipidosis, as well as certain apoptotic-associated events (caspase activation, lysosomal degradation) (30,41), it is tempting to speculate that phospholipidosis and cell death may have common signaling pathways. Therefore, the ability of cytotoxic oxysterols to activate intracellular lipid accumulation (mainly polar lipids) favors the hypothesis that the balance between these oxysterols and vitamin E might play key roles in the lipid profile of atherosclerotic lesions (Figure 3), and subsequently in their progression since this latter depends on the biological activities of the lipids considered (59). Interestingly, in agreement with these in vitro observations, increased concentrations of oxysterols measured in the plasma from coronary artery bypass graft patients were associated with a substantial enhancement of sphingomyelin in coronary artery tissue (60).
Conclusions
The present review underlines the multiple activities and mechanisms of oxysterols and the complexity of their action. As judged from the results of animal experiments, the involvement of oxysterols in atherogenesis is highly controversial but cannot be excluded. Consequently, more appropriate in vivo and in vitro models of investigation as well as clinical investigations are needed to clarify the debate. However, although the role of oxysterols in atherosclerosis remains to be elucidated, the large number of different biological activities of these molecules, the presence of putative oxysterol receptors in various cell types, and/or the similarities between atherosclerosis and other degenerative diseases such as Alzheimer's disease, osteoporosis, and age-related macular degeneration has stimulated new investigations. Indeed, as data obtained on rat hippocampal H19-7/IGF-IR cells have revealed marked cytotoxic effects of 7β-hydroxycholesterol in concentrations ranging from 10 to 100 nM (9), instead of 30 to 100 µM in the cells of the vascular wall and in monocytic cells (U937 and THP-1 cells) (15,33), better knowledge of oxysterol activities still remains an important and essential source of investigation that can contribute to improving our understanding in various major pathologies and to developing new and efficient treatments.
Structures of cholesterol and major oxysterols. Oxysterols are cholesterol oxide products resulting from the autoxidation or enzymatic oxidation of cholesterol. The major oxysterols resulting from autoxidation (7-ketocholesterol; 7α-hydroxycholesterol, and its enantiomer 7β-hydroxycholesterol) and enzymatic oxidation (cholesterol-5α, 6α-epoxide, and its enantiomer cholesterol-5β,6β-epoxide) of cholesterol are represented.
Signaling pathways associated with 7-ketocholesterol- and 7β-hydroxycholesterol-induced apoptosis. 7-Ketocholesterol- and 7β-hydroxycholesterol-induced cell death are characterized by an early externalization of phosphatidylserine and by the following events: Ca2+ influx; activation of calmodulin and calcineurin leading to Bad dephosphorylation and subsequent mitochondrial depolarization (loss of transmembrane mitochondrial potential ΔΨm); mitochondrial release of cytochrome c, apoptosis-inducing factor (AIF) and endonuclease-G (Endo-G) into the cytosol; caspase-3, -7, -8, and -9 activation; Bid truncation; poly(ADP-ribose)polymerase (PARP) degradation; cleavage of the DNA fragmentation factor (DFF45)/inhibitor of caspase-activated DNase (ICAD), leading to the activation of caspase-activated DNase (CAD) involved in internucleosomal DNA fragmentation. The ability of 7-ketocholesterol and 7β-hydroxycholesterol to induce an overproduction of reactive oxygen species (ROS) contributes to decrease the transmembrane mitochondrial potential (ΔΨm) and the intracellular level of reduced glutathione (GSH). In addition, 7-ketocholesterol inhibits the PDK1/Akt (PKB) signaling pathway, and both 7-ketocholesterol and 7β-hydroxycholesterol trigger the formation of multilamellar cytoplasmic structures (myelin figures) containing high levels of phospholipids. (Copyright Dr. Gérard Lizard, Inserm, France).
Effects of vitamin-E on 7-ketocholesterol-, and 7β-hydroxycholesterol-induced cell death. When U937, THP-1, or vascular cells are cultured in the presence of 7-ketocholesterol or 7β-hydroxycholesterol, cell death is associated with nuclear morphological changes. Some cells with swollen nuclei (oncotic cells), as well as with condensed and/or fragmented nuclei, are simultaneously detected. Moreover, under treatment with 7-ketocholesterol and 7β-hydroxycholesterol, myelin figures and polar lipid accumulation are observed as well as an overproduction of reactive oxygen species (ROS). In vivo, these events could give foam cells (lipid-laden macrophages) known to contribute to the early steps of the development of atherosclerotic lesions (formation of the fatty streak) and probably also to the formation of the lipid core in more advanced lesions. Vitamin E (Vit-E), which is able to counteract 7-ketocholesterol- and 7β-hydroxycholesterol-induced apoptosis, overproduction of ROS, myelin figure formation and phospholipidosis, could consequently contribute to reducing the release of some cellular components from dying cells (phospholipids, cholesterol, oxysterols) in the arterial wall, and therefore inhibit the formation of the lipid core and reduce plaque instability. (Copyright Dr. Gérard Lizard, Inserm, France).
References
Acknowledgments
The authors are indebted to Ms. Linda Northrup for reviewing the English version of the manuscript.
Address for correspondence: G. Lizard, Faculté des Sciences Gabriel, Centre de Recherche Inserm U866, Equipe Biochimie Métabolique et Nutritionnelle, 6 Bd Gabriel, 21000 Dijon, France. Fax: +33-380-39-6250. E-mail: gerard.lizard@u-bourgogne.fr
Research supported by grants from the INSERM, the Ligue Contre Le Cancer (Comité de Côte d'Or), the University Hospital of Dijon, and the Conseil Régional de Bourgogne. Presented at the XXII Annual Meeting of the Federação de Sociedades de Biologia Experimental, Águas de Lindóia, SP, Brazil, August 22-25, 2007. Received March 24, 2008. Accepted July 4, 2008.
- 1. Guardiola F, Dutta PC, Codony R, Savage GP. Cholesterol and phytosterol oxydation products: analysis, occurrence, and biological effects Champaign: AOCS Press; 2002.
- 2. Smith LL. Mechanisms of formation of oxysterols: a general survey. In: Crastes de Paulet A, Douste-Blazy L, Paoletti R (Editors), Free radicals, lipoproteins, and membrane lipids New York: Plenum Press; 1990. p 115-132.
- 3. Girotti AW. Translocation as a means of disseminating lipid hydroperoxide-induced oxidative damage and effector action. Free Radic Biol Med 2008; 44: 956-968.
- 4. Schweizer RA, Zurcher M, Balazs Z, Dick B, Odermatt A. Rapid hepatic metabolism of 7-ketocholesterol by 11beta-hydroxysteroid dehydrogenase type 1: species-specific differences between the rat, human, and hamster enzyme. J Biol Chem 2004; 279: 18415-18424.
- 5. Colles SM, Maxson JM, Carlson SG, Chisolm GM. Oxidized LDL-induced injury and apoptosis in atherosclerosis. Potential roles for oxysterols. Trends Cardiovasc Med 2001; 11: 131-138.
- 6. Bretillon L, Diczfalusy U, Bjorkhem I, Maire MA, Martine L, Joffre C, et al. Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr Eye Res 2007; 32: 361-366.
- 7. Björkhem I. Mechanism of degradation of the steroid side chain in the formation of bile acids. J Lipid Res 1992; 33: 455-471.
- 8. Maeda Y, Nagatomo H, Uchiyama F, Nagatomo J, Yamada M, Shiotsuki H, et al. A comparative study of the conversion of 7-hydroxycholesterol in rabbit, guinea pig, rat, hamster, and chicken. Steroids 2002; 67: 703-708.
- 9. Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J Biol Chem 2005; 280: 7377-7387.
- 10. Wentworth P Jr, Nieva J, Takeuchi C, Galve R, Wentworth AD, Dilley RB, et al. Evidence for ozone formation in human atherosclerotic arteries. Science 2003; 302: 1053-1056.
- 11. Smith LL. Oxygen, oxysterols, ouabain, and ozone: a cautionary tale. Free Radic Biol Med 2004; 37: 318-324.
- 12. Sathishkumar K, Murthy SN, Uppu RM. Cytotoxic effects of oxysterols produced during ozonolysis of cholesterol in murine GT1-7 hypothalamic neurons. Free Radic Res 2007; 41: 82-88.
- 13. Schroepfer GJ Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev 2000; 80: 361-554.
- 14. Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis 1999; 142: 1-28.
- 15. Lemaire-Ewing S, Prunet C, Montange T, Vejux A, Berthier A, Bessede G, et al. Comparison of the cytotoxic, pro-oxidant and pro-inflammatory characteristics of different oxysterols. Cell Biol Toxicol 2005; 21: 97-114.
- 16. Hayden JM, Brachova L, Higgins K, Obermiller L, Sevanian A, Khandrika S, et al. Induction of monocyte differentiation and foam cell formation in vitro by 7-ketocholesterol. J Lipid Res 2002; 43: 26-35.
- 17. Malvitte L, Montange T, Joffre C, Vejux A, Maiza C, Bron A, et al. Analogies between atherosclerosis and age-related maculopathy: expected roles of oxysterols. J Fr Ophtalmol 2006; 29: 570-578.
- 18. Cao G, Bales KR, DeMattos RB, Paul SM. Liver X receptor-mediated gene regulation and cholesterol homeostasis in brain: relevance to Alzheimer's disease therapeutics. Curr Alzheimer Res 2007; 4: 179-184.
- 19. Liu H, Yuan L, Xu S, Wang K, Zhang T. Cholestane-3beta,5alpha,6beta-triol inhibits osteoblastic differentiation and promotes apoptosis of rat bone marrow stromal cells. J Cell Biochem 2005; 96: 198-208.
- 20. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 2005; 73: 1907-1916.
- 21. Assunção GC, Linden R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur J Biochem 2004; 271: 1638-1650.
- 22. Ogier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003; 1603: 113-128.
- 23. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717-1721.
- 24. Prunet C, Lemaire-Ewing S, Menetrier F, Neel D, Lizard G. Activation of caspase-3-dependent and -independent pathways during 7-ketocholesterol- and 7beta-hydroxycholesterol-induced cell death: a morphological and biochemical study. J Biochem Mol Toxicol 2005; 19: 311-326.
- 25. Lizard G, Monier S, Cordelet C, Gesquiere L, Deckert V, Gueldry S, et al. Characterization and comparison of the mode of cell death, apoptosis versus necrosis, induced by 7beta-hydroxycholesterol and 7-ketocholesterol in the cells of the vascular wall. Arterioscler Thromb Vasc Biol 1999; 19: 1190-1200.
- 26. Lizard G, Deckert V, Dubrez L, Moisant M, Gambert P, Lagrost L. Induction of apoptosis in endothelial cells treated with cholesterol oxides. Am J Pathol 1996; 148: 1625-1638.
- 27. Miguet-Alfonsi C, Prunet C, Monier S, Bessede G, Lemaire-Ewing S, Berthier A, et al. Analysis of oxidative processes and of myelin figures formation before and after the loss of mitochondrial transmembrane potential during 7beta-hydroxycholesterol and 7-ketocholesterol-induced apoptosis: comparison with various pro-apoptotic chemicals. Biochem Pharmacol 2002; 64: 527-541.
- 28. Martinet W, De Bie M, Schrijvers DM, De Meyer GR, Herman AG, Kockx MM. 7-Ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2004; 24: 2296-2301.
- 29. Anderson N, Borlak J. Drug-induced phospholipidosis. FEBS Lett 2006; 580: 5533-5540.
- 30. Vejux A, Kahn E, Menetrier F, Montange T, Lherminier J, Riedinger JM, et al. Cytotoxic oxysterols induce caspase-independent myelin figure formation and caspase-dependent polar lipid accumulation. Histochem Cell Biol 2007; 127: 609-624.
- 31. Lizard G, Moisant M, Cordelet C, Monier S, Gambert P, Lagrost L. Induction of similar features of apoptosis in human and bovine vascular endothelial cells treated by 7-ketocholesterol. J Pathol 1997; 183: 330-338.
- 32. Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol 2004; 24: 10703-10717.
- 33. Berthier A, Lemaire-Ewing S, Prunet C, Monier S, Athias A, Bessede G, et al. Involvement of a calcium-dependent dephosphorylation of BAD associated with the localization of Trpc-1 within lipid rafts in 7-ketocholesterol-induced THP-1 cell apoptosis. Cell Death Differ 2004; 11: 897-905.
- 34. Han JH, Kim YJ, Han ES, Lee CS. Prevention of 7-ketocholesterol-induced mitochondrial damage and cell death by calmodulin inhibition. Brain Res 2007; 1137: 11-19.
- 35. Panini SR, Yang L, Rusinol AE, Sinensky MS, Bonventre JV, Leslie CC. Arachidonate metabolism and the signaling pathway of induction of apoptosis by oxidized LDL/oxysterol. J Lipid Res 2001; 42: 1678-1686.
- 36. Lee T, Chau L. Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells. Am J Physiol Cell Physiol 2001; 280: C709-C718.
- 37. Rho MC, Kim YK, Chang JS, Lee HS, Baek JA, Chung MY, et al. 7-Ketocholesterol predisposes human aorta smooth muscle cells to Fas-mediated death. J Mol Cell Cardiol 2005; 39: 823-832.
- 38. Vejux A, Lizard G, Tourneur Y, Riedinger JM, Frouin F, Kahn E. Effects of caspase inhibitors (z-VAD-fmk, z-VDVAD-fmk) on Nile Red fluorescence pattern in 7-ketocholesterol-treated cells: investigation by flow cytometry and spectral imaging microscopy. Cytometry A 2007; 71: 550-562.
- 39. Miguet C, Monier S, Bettaieb A, Athias A, Bessede G, Laubriet A, et al. Ceramide generation occurring during 7beta-hydroxycholesterol- and 7-ketocholesterol-induced apoptosis is caspase independent and is not required to trigger cell death. Cell Death Differ 2001; 8: 83-99.
- 40. Lordan S, O'Neill C, O'Brien NM. Effects of apigenin, lycopene and astaxanthin on 7beta-hydroxycholesterol-induced apoptosis and Akt phosphorylation in U937 cells. Br J Nutr 2008; 1-10.
- 41. Lizard G, Miguet C, Bessede G, Monier S, Gueldry S, Neel D, et al. Impairment with various antioxidants of the loss of mitochondrial transmembrane potential and of the cytosolic release of cytochrome c occuring during 7-ketocholesterol-induced apoptosis. Free Radic Biol Med 2000; 28: 743-753.
- 42. Biasi F, Leonarduzzi G, Vizio B, Zanetti D, Sevanian A, Sottero B, et al. Oxysterol mixtures prevent proapoptotic effects of 7-ketocholesterol in macrophages: implications for proatherogenic gene modulation. FASEB J 2004; 18: 693-695.
- 43. Larsson DA, Baird S, Nyhalah JD, Yuan XM, Li W. Oxysterol mixtures, in atheroma-relevant proportions, display synergistic and proapoptotic effects. Free Radic Biol Med 2006; 41: 902-910.
- 44. Ringseis R, Eder K. Dietary oxidized cholesterol increases expression and activity of antioxidative enzymes and reduces the concentration of glutathione in the liver of rats. Int J Vitam Nutr Res 2004; 74: 86-92.
- 45. Rosenblat M, Aviram M. Oxysterol-induced activation of macrophage NADPH-oxidase enhances cell-mediated oxidation of LDL in the atherosclerotic apolipoprotein E deficient mouse: inhibitory role for vitamin E. Atherosclerosis 2002; 160: 69-80.
- 46. Lizard G, Gueldry S, Sordet O, Monier S, Athias A, Miguet C, et al. Glutathione is implied in the control of 7-ketocholesterol-induced apoptosis, which is associated with radical oxygen species production. FASEB J 1998; 12: 1651-1663.
- 47. Monier S, Samadi M, Prunet C, Denance M, Laubriet A, Athias A, et al. Impairment of the cytotoxic and oxidative activities of 7 beta-hydroxycholesterol and 7-ketocholesterol by esterification with oleate. Biochem Biophys Res Commun 2003; 303: 814-824.
- 48. O'Sullivan AJ, O'Callaghan YC, O'Brien NM. Differential effects of mixtures of cholesterol oxidation products on bovine aortic endothelial cells and human monocytic U937 cells. Int J Toxicol 2005; 24: 173-179.
- 49. Liu SX, Chen Y, Zhou M, Wan J. Oxidized cholesterol in oxidized low density lipoprotein may be responsible for the inhibition of LPS-induced nitric oxide production in macrophages. Atherosclerosis 1998; 136: 43-49.
- 50. Shouhed D, Kha HT, Richardson JA, Amantea CM, Hahn TJ, Parhami F. Osteogenic oxysterols inhibit the adverse effects of oxidative stress on osteogenic differentiation of marrow stromal cells. J Cell Biochem 2005; 95: 1276-1283.
- 51. Rydberg EK, Salomonsson L, Hulten LM, Noren K, Bondjers G, Wiklund O, et al. Hypoxia increases 25-hydroxycholesterol-induced interleukin-8 protein secretion in human macrophages. Atherosclerosis 2003; 170: 245-252.
- 52. Leonarduzzi G, Gamba P, Sottero B, Kadl A, Robbesyn F, Calogero RA, et al. Oxysterol-induced up-regulation of MCP-1 expression and synthesis in macrophage cells. Free Radic Biol Med 2005; 39: 1152-1161.
- 53. Leonarduzzi G, Sevanian A, Sottero B, Arkan MC, Biasi F, Chiarpotto E, et al. Up-regulation of the fibrogenic cytokine TGF-beta1 by oxysterols: a mechanistic link between cholesterol and atherosclerosis. FASEB J 2001; 15: 1619-1621.
- 54. Prunet C, Montange T, Vejux A, Laubriet A, Rohmer JF, Riedinger JM, et al. Multiplexed flow cytometric analyses of pro- and anti-inflammatory cytokines in the culture media of oxysterol-treated human monocytic cells and in the sera of atherosclerotic patients. Cytometry A 2006; 69: 359-373.
- 55. Antonio V, Janvier B, Brouillet A, Andreani M, Raymondjean M. Oxysterol and 9-cis-retinoic acid stimulate the group IIA secretory phospholipase A2 gene in rat smooth-muscle cells. Biochem J 2003; 376: 351-360.
- 56. Englund MC, Karlsson AL, Wiklund O, Bondjers G, Ohlsson BG. 25-Hydroxycholesterol induces lipopolysaccharide-tolerance and decreases a lipopolysaccharide-induced TNF-alpha secretion in macrophages. Atherosclerosis 2001; 158: 61-71.
- 57. Piccotti JR, LaGattuta MS, Knight SA, Gonzales AJ, Bleavins MR. Induction of apoptosis by cationic amphiphilic drugs amiodarone and imipramine. Drug Chem Toxicol 2005; 28: 117-133.
- 58. Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol 2003; 5: 781-792.
- 59. Ninio E. Phospholipid mediators in the vessel wall: involvement in atherosclerosis. Curr Opin Clin Nutr Metab Care 2005; 8: 123-131.
- 60. Kummerow FA, Cook LS, Wasowicz E, Jelen H. Changes in the phospholipid composition of the arterial cell can result in severe atherosclerotic lesions. J Nutr Biochem 2001; 12: 602-607.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
14 Aug 2008 -
Date of issue
July 2008
History
-
Accepted
04 July 2008 -
Received
24 Mar 2008