Abstract
Male sex determination in humans is controlled by the SRY gene, which encodes a transcriptional regulator containing a conserved high mobility group box domain (HMG-box) required for DNA binding. Mutations in the SRY HMG-box affect protein function, causing sex reversal phenotypes. In the present study, we describe a 19-year-old female presenting 46,XY karyotype with hypogonadism and primary amenorrhea that led to the diagnosis of 46,XY complete gonadal dysgenesis. The novel p.E89K missense mutation in the SRY HMG-box was identified as a de novo mutation. Electrophoretic mobility shift assays showed that p.E89K almost completely abolished SRY DNA-binding activity, suggesting that it is the cause of SRY function impairment. In addition, we report the occurrence of the p.G95R mutation in a 46,XY female with complete gonadal dysgenesis. According to the three-dimensional structure of the human SRY HMG-box, the substitution of the conserved glutamic acid residue by the basic lysine at position 89 introduces an extra positive charge adjacent to and between the positively charged residues R86 and K92, important for stabilizing the HMG-box helix 2 with DNA. Thus, we propose that an electrostatic repulsion caused by the proximity of these positive charges could destabilize the tip of helix 2, abrogating DNA interaction.
Gonadal dysgenesis; HMG-box; Missense mutation; Sex reversal SRY; Streak gonads; Testis determination
Braz J Med Biol Res, April 2010, Volume 44(4) 361-365
The novel p.E89K mutation in the SRY gene inhibits DNA binding and causes the 46,XY disorder of sex development
J.L. Cunha1, F.C. Soardi1, R.D. Bernardi1, L.E.C. Oliveira1, C.E. Benedetti2, G. Guerra-Junior3,4, A.T. Maciel-Guerra3,5 and  Correspondence and Footnotes
Correspondence and Footnotes
 M.P. de Mello1,3
M.P. de Mello1,3
1Centro de Biologia Molecular e Engenharia Genética, Universidade Estadual de Campinas, Campinas, SP, Brasil
2Laboratório Nacional de Biociências, Centro Nacional de Pesquisa em Energia e Materiais, Campinas, SP, Brasil
3Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo, 4Departamento de Pediatria, 5Departamento de Genética Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Campinas, SP, Brasil
 Patients and Methods
Patients and Methods
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Male sex determination in humans is controlled by the SRY gene, which encodes a transcriptional regulator containing a conserved high mobility group box domain (HMG-box) required for DNA binding. Mutations in the SRY HMG-box affect protein function, causing sex reversal phenotypes. In the present study, we describe a 19-year-old female presenting 46,XY karyotype with hypogonadism and primary amenorrhea that led to the diagnosis of 46,XY complete gonadal dysgenesis. The novel p.E89K missense mutation in the SRY HMG-box was identified as a de novo mutation. Electrophoretic mobility shift assays showed that p.E89K almost completely abolished SRY DNA-binding activity, suggesting that it is the cause of SRY function impairment. In addition, we report the occurrence of the p.G95R mutation in a 46,XY female with complete gonadal dysgenesis. According to the three-dimensional structure of the human SRY HMG-box, the substitution of the conserved glutamic acid residue by the basic lysine at position 89 introduces an extra positive charge adjacent to and between the positively charged residues R86 and K92, important for stabilizing the HMG-box helix 2 with DNA. Thus, we propose that an electrostatic repulsion caused by the proximity of these positive charges could destabilize the tip of helix 2, abrogating DNA interaction.
Key words: Gonadal dysgenesis; HMG-box; Missense mutation; Sex reversal SRY; Streak gonads; Testis determination
Introduction
In humans, as in other mammals, sex determination is controlled by a dominant switch located on the Y-chromosome, the SRY gene, also referred to as the sex-determining region on the Y (1,2). The human single copy monoexonic SRY gene encodes a 204-amino acid protein that contains a conserved domain formed by 79 residues known as the high mobility group box (HMG-box), which is flanked by two nuclear localization signals (3). The HMG-box characterizes the SRY as a transcription regulator with DNA-binding and bending abilities (4-6). Mutations within the SRY gene sequence cause male-to-female sex reversal in XY individuals as a result of failure of indifferent gonads to develop into testes, leading to complete XY gonadal dysgenesis (46,XY CGD) (7), included among the 46,XY disorders of sex development (8). Patients with 46,XY CGD present a normal female phenotype and streak gonads at birth, whereas they will present primary amenorrhea and failure to develop female secondary characteristics at puberty.
The importance of SRY function has been indicated by a number of studies describing amino acid changes within this protein in patients with 46,XY CGD (9). To date, up to 68 mutations have been identified within the open reading frame of the SRY gene (10). The majority of these mutations are located within the HMG-box, highlighting the critical role of this domain in DNA binding and transcriptional activation (11). Although mutations outside the SRY HMG-box domain are rare, they can also cause 46,XY CGD (12,13). Furthermore, a small subset of reports describe familial mutations (13,14), whereas the majority are de novo mutations affecting only one individual in a family. In summary, a large spectrum of phenotypic variation is observed among patients carrying different SRY gene mutations; therefore, phenotype and genotype correlations in patients with XY gonadal dysgenesis are not straightforward.
In the present study, the novel p.E89K and p.G95R mutations within the SRY HMG-box were identified in patients with 46,XY CGD. The E89 residue is conserved among the SRY proteins and the p.E89K mutation significantly affects the protein DNA-binding activity in vitro. Interestingly, both mutations introduce an extra positive charge adjacent to and between the positively charged residues R86 and K92 important for stabilizing the HMG-box helix 2 and DNA complex. The role of the electrostatic repulsion caused by the proximity of those positive charges on DNA interaction is discussed.
Patients and Methods

Patients
Patient 1. A 19-year-old female was referred to us due to hypogonadism and primary amenorrhea. She was born at term by normal delivery, but her birth weight is unknown. She was the first child of non-consanguineous parents in a sibship of 8 (6 sisters and 1 brother) with unremarkable family history. Upon physical examination, her weight was 42.4 kg and height 158 cm. She had female external genitalia with pubertal development in Tanner stage M1P2, and there was no dysmorphic picture. Cytogenetic analysis revealed a 46,XY karyotype. There were high levels of FSH (164 mIU/mL; normal range (NR): 1.5-12.4 mIU/mL) and LH (31.1 mIU/mL; NR: 1.7-8.6 mIU/mL), and ultrasonography revealed infantile uterus, but no female gonads were visualized. Both dysgenetic gonads were removed and a microscopic gonadoblastoma (2 x 2 mm) was found in the right gonad.
Patient 2. A 17-year-old female was referred to us due to hypogonadism and primary amenorrhea. She was born at term by normal delivery to nonconsanguineous parents, with 2.6 kg and 46 cm of birth weight and length, respectively. Her mother had a stillborn girl and a first-trimester miscarriage, followed by an 18-year-old girl and the proposita; family history was unremarkable. On physical examination, her weight was 67.8 kg and height 174.3 cm. She had female external genitalia with pubertal development in Tanner stage M1P3, and no dysmorphic characteristics. Cytogenetic analysis revealed a 46,XY karyotype. There were high levels of FSH (80.1 mIU/mL; NR: 1.5-12.4 mIU/mL) and LH (21.4 mIU/mL; NR: 1.7-8.6 mIU/mL), and ultrasonography revealed infantile uterus, but no female gonads were visualized. The patient was informed about the risk of malignant development of gonadal tissue and was referred to other services in order to perform prophylactic bilateral gonadectomy.
The study was approved by the Ethics Committee of the Universidade Estadual de Campinas, and informed consent was obtained from the patients and controls.
Methods
Genomic DNA was extracted from peripheral blood by standard techniques (15). PCR was performed with 0.5 µg genomic DNA, 10% DMSO, 200 µM each deoxyribonucleoside triphosphates, 0.5 µM each primer, 1X Taq DNA polymerase buffer solution (Amersham-GE, USA), and 1.5 U Taq DNA polymerase (Invitrogen, USA). Each PCR contained normal female and male control DNA. The SRY open reading frame was amplified as described before (13) and sequenced twice with products from different PCR assays. Both sense and antisense primers were used to confirm sequence results. Amplified SRY fragments were purified with Wizard SV Gel and PCR Clean-UP System (Promega, USA) and directly sequenced with the Big Dye Terminator Cycle Sequencing Kit V3.1 Ready Reaction (Applied Biosystems, USA). Wild-type and mutant SRY gene samples were amplified by PCR to be cloned into pET28A bacterial expression vector (Novagen, USA) to produce N-terminal His-tagged fusion proteins as described elsewhere (13). Resulting constructs were used to transform BL21 (DE3) competent Escherichia coli cells, and the expression of wild-type and mutant SRY proteins was induced with 0.1 mM isopropyl-1-thio-beta-D-galactopyranoside (IPTG). Cells were collected and lysed and protein was purified with an Ni-NTA agarose column (Qiagen, Germany). SRY proteins were eluted and stored at -70°C. Specific oligonucleotides containing the SRY-target DNA sequence were synthesized and subsequently annealed to generate a double-stranded probe (oligoA) as previously described (13,16). The ds-oligonucleotide was labeled with (α-32P) dCTP (Amersham-GE) using the standard Klenow fill-in reaction. The mixtures were incubated in the presence of increasing concentrations of either normal or mutant SRY proteins. Electrophoretic mobility shift assays (EMSAs) were carried out on non-denaturing 6% polyacrylamide gels and the shifted bands were visualized on Hiperfilm MP autoradiographies (Amersham-GE).
Results
The 380-bp amplified fragment encoding the SRY conserved domain was sequenced with 46,XY CGD in both female patients. A hemizygous transition of a guanine (GAG) to an adenine (AAG) in codon 89 was observed in patient 1 (Figure 1A). This novel mutation causes the substitution of a glutamic acid by a lysine (p.E89K) within the HMG-box of the SRY gene. Sequencing the whole SRY coding region did not show any other nucleotide variation. The patient’s father and brother did not present the nucleotide change, suggesting that this mutation had occurred as a de novo event. The p.G95R mutation was identified in patient 2 as a result of the GGA>CGA nucleotide change (Figure 1B).
To investigate the effect of the p.E89K mutation on the SRY DNA-binding activity, EMSAs were performed with the purified wild-type and mutant proteins. The assay with increasing protein concentrations showed that the mutant protein was unable to bind to the DNA probe even at high concentrations, as compared to the wild-type protein, indicating that the mutation drastically decreased the SRY DNA-binding activity (Figure 1C).
Partial electropherograms of SRY gene DNA sequences of normal control and patients with 46,XY complete gonadal dysgenesis are shown. A, Patient 1 showing the G>A substitution that leads to a change in the amino acid E89 to K, within the highly conserved DNA-binding motif (HMG-box) of the SRY protein. B, Patient 2 showing the G>C substitution that leads to a change in the amino acid G95 to R, within the highly conserved DNA-binding motif (HMG-box) of the SRY protein. C, Effect of the p.E89K mutation on the DNA-binding activity of the SRY protein. Electrophoretic mobility shift assays (EMSA) were performed using the purified wild-type and mutant SRY proteins. Lane 1 corresponds to the 32P-labeled free probe. Lanes 2, 4, 6, and 8 correspond to reactions comprising the probe + wild-type SRY protein at increasing concentrations (0.5, 1, 2.5, and 5 µg, respectively). The arrow indicates the SRY-DNA complex. Lanes 3, 5, 7, and 9 correspond to reactions containing the probe + mutant SRYE89K protein at increasing concentrations (0.5, 1, 2.5, and 5 µg, respectively).
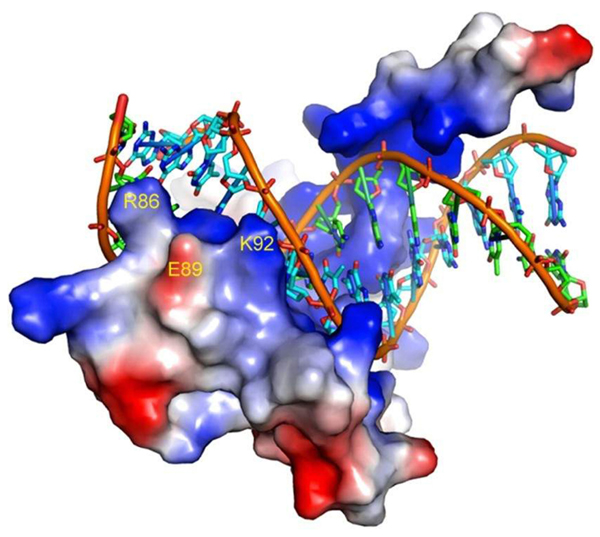
Representation of the SRY-DNA complex depicting the tip of helix 2 involved in the DNA interaction. The electrostatic surface of SRY shows that the side chain of E89 sits between R86 and K92. The picture was created with the program PyMOL using the 3-D coordinates of the SRY protein (PDB code 1J46) published by Murphy et al. (21).
Discussion
In the present study, we report a case of p.G95R substitution associated with sex reversal and describe a novel mutation (p.E89K) within the SRY HMG-box in patients with 46,XY pure gonadal dysgenesis. The p.G95R mutation has already been described in cases of complete XY gonadal dysgenesis and has been shown to affect the DNA-binding activity of the protein (17-19).
The majority of the SRY mutations reported in patients with diseases of sex differentiation occur within the HMG-box domain (20). Not surprisingly, we found that the p.E89K mutation also impaired significantly the in vitro DNA-binding activity of the SRY protein. Although the E89 residue does not make contacts with DNA (21), its replacement by a positively charged residue is likely to have caused alterations in the electrostatic net charge of the protein surface, since E89 is located outside the HMG-box helix 2 (21). In addition, it is worth noting that in the 3-D structure of SRY (Protein Data Bank - PDB code 1J46), the side chain of E89 is located adjacent to and between the side chains of residues R86 and K92, both of which stabilize helix 2 through hydrogen bonds with the phosphate groups of DNA (see Ref. 21 and Figure 2). Since in the protein-DNA complex the atomic distances between E89 and the adjacent R86 and K92 side chains are outside the normal range for a strong electrostatic interaction (>4 Å), E89 may play a critical role to hold both R86 and K92 side chains in position to interact with DNA when the protein is not bound to its target sequence. Therefore, it is reasonable to suggest that the insertion of an extra positive charge between R86 and K92 would cause strong electrostatic repulsion among the side chains leading to the destabilization of the tip of helix 2, precluding DNA interaction. The fact that E89 is conserved in SRY protein in various mammalian species (22) is a strong indication that an acidic side chain in this position is important to maintain the protein function. Mutations in the same region of the SRY protein involving residues 90, 91, 94, and 95 also produced proteins with reduced binding activity (18). Accordingly, the G to R change at position 95 reported here also places a strong positive charge in the region adjacently to the K92 side chain. However, in this case, the insertion of an R residue might favor an inadvertent contact with the phosphate group, breaking hydrogen bonds formed by the R86 and K92 side chains with the DNA.
In conclusion, there is a good correlation between in vitro and in vivo effects involving mutant SRY proteins identified in the present report.
References
1. Berta P, Hawkins JR, Sinclair AH, Taylor A, Griffiths BL, Goodfellow PN, et al. Genetic evidence equating SRY and the testis-determining factor. Nature 1990; 348: 448-450.
2. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990; 346: 240-244.
3. Behlke MA, Bogan JS, Beer-Romero P, Page DC. Evidence that the SRY protein is encoded by a single exon on the human Y chromosome. Genomics 1993; 17: 736-739.
4. Harley VR, Jackson DI, Hextall PJ, Hawkins JR, Berkovitz GD, Sockanathan S, et al. DNA binding activity of recombinant SRY from normal males and XY females. Science 1992; 255: 453-456.
5. Pontiggia A, Rimini R, Harley VR, Goodfellow PN, Lovell-Badge R, Bianchi ME. Sex-reversing mutations affect the architecture of SRY-DNA complexes. EMBO J 1994; 13: 6115-6124.
6. Giese K, Pagel J, Grosschedl R. Distinct DNA-binding properties of the high mobility group domain of murine and human SRY sex-determining factors. Proc Natl Acad Sci USA 1994; 91: 3368-3372.
7. Gimelli G, Gimelli S, Dimasi N, Bocciardi R, Di Battista E, Pramparo T, et al. Identification and molecular modelling of a novel familial mutation in the SRY gene implicated in the pure gonadal dysgenesis. Eur J Hum Genet 2007; 15: 76-80.
8. Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Arch Dis Child 2006; 91: 554-563.
9. Knower KC, Kelly S, Harley VR. Turning on the male - SRY, SOX9 and sex determination in mammals. Cytogenet Genome Res 2003; 101: 185-198.
10. Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, et al. The Human Gene Mutation Database: 2008 update. Genome Med 2009; 1: 13.
11. Shahid M, Dhillion VS, Jain N, Hedau S, Diwakar S, Sachdeva P, et al. Two new novel point mutations localized upstream and downstream of the HMG box region of the SRY gene in three Indian 46,XY females with sex reversal and gonadal tumour formation. Mol Hum Reprod 2004; 10: 521-526.
12. Marchina E, Gambera A, Spinelli E, Clerici P, Scagliola P, Sartori E, et al. Identification of a new mutation in the SRY gene in a 46,XY woman with Swyer syndrome. Fertil Steril 2009; 91: 932.
13. Assumpcao JG, Benedetti CE, Maciel-Guerra AT, Guerra G Jr, Baptista MT, Scolfaro MR, et al. Novel mutations affecting SRY DNA-binding activity: the HMG box N65H associated with 46,XY pure gonadal dysgenesis and the familial non-HMG box R30I associated with variable phenotypes. J Mol Med 2002; 80: 782-790.
14. Plaseska-Karanfilska D, Noveski P, Kuzevska K, Basheska N, Kocova M, Efremov GD. A new familial mutation (R133G) in the SRY gene. Clin Genet 2007; 71: 480-482.
15. Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3rd edn. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001.
16. Schmitt-Ney M, Thiele H, Kaltwasser P, Bardoni B, Cisternino M, Scherer G. Two novel SRY missense mutations reducing DNA binding identified in XY females and their mosaic fathers. Am J Hum Genet 1995; 56: 862-869.
17. Hawkins JR, Taylor A, Goodfellow PN, Migeon CJ, Smith KD, Berkovitz GD. Evidence for increased prevalence of SRY mutations in XY females with complete rather than partial gonadal dysgenesis. Am J Hum Genet 1992; 51: 979-984.
18. Rimini R, Pontiggia A, Spada F, Ferrari S, Harley VR, Goodfellow PN, et al. Interaction of normal and mutant SRY proteins with DNA. Philos Trans R Soc Lond B Biol Sci 1995; 350: 215-220.
19. Copelli SB, Pasqualini T. Association of Turner’s syndrome and Swyer’s syndrome in the same family. J Pediatr Endocrinol Metab 2000; 13: 557-559.
20. Harley VR, Clarkson MJ, Argentaro A. The molecular action and regulation of the testis-determining factors, SRY (sex-determining region on the Y chromosome) and SOX9 [SRY-related high-mobility group (HMG) box 9]. Endocr Rev 2003; 24: 466-487.
21. Murphy EC, Zhurkin VB, Louis JM, Cornilescu G, Clore GM. Structural basis for SRY-dependent 46-X,Y sex reversal: modulation of DNA bending by a naturally occurring point mutation. J Mol Biol 2001; 312: 481-499.
22. Shahid M, Dhillon VS, Hussain Z, Masa JF, Aslam M, Raish M, et al. Analysis of the SRY gene in two sex-reversed XY sisters identifies two new novel point mutations in the high mobility group box domain. Fertil Steril 2008; 90: 1199.e1-8.
Acknowledgments
Research supported by FAPESP (#08/54776-1 to M.P. de Mello, and scholarships #02/13237-4, #07/00900-0, and #08/03168-1 to L.E.C. Oliveira, J.L. Cunha and F.C. Soardi, respectively. We also thank CAPES and CNPq.
Address for correspondence: M.P. de Mello, CBMEG-UNICAMP, Caixa Postal 6010, 13083-875 Campinas, SP, Brasil. Fax: +55-19-3521-1089. E-mail: mmello@unicamp.br
Received October 7, 2010. Accepted January 31, 2011. Available online February 11, 2011. Published April 11, 2011.
The Brazilian Journal of Medical and Biological Research is partially financed by
- 1. Berta P, Hawkins JR, Sinclair AH, Taylor A, Griffiths BL, Goodfellow PN, et al. Genetic evidence equating SRY and the testis-determining factor. Nature 1990; 348: 448-450.
- 2. Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990; 346: 240-244.
- 3. Behlke MA, Bogan JS, Beer-Romero P, Page DC. Evidence that the SRY protein is encoded by a single exon on the human Y chromosome. Genomics 1993; 17: 736-739.
- 4. Harley VR, Jackson DI, Hextall PJ, Hawkins JR, Berkovitz GD, Sockanathan S, et al. DNA binding activity of recombinant SRY from normal males and XY females. Science 1992; 255: 453-456.
- 5. Pontiggia A, Rimini R, Harley VR, Goodfellow PN, Lovell-Badge R, Bianchi ME. Sex-reversing mutations affect the architecture of SRY-DNA complexes. EMBO J 1994; 13: 6115-6124.
- 6. Giese K, Pagel J, Grosschedl R. Distinct DNA-binding properties of the high mobility group domain of murine and human SRY sex-determining factors. Proc Natl Acad Sci USA 1994; 91: 3368-3372.
- 7. Gimelli G, Gimelli S, Dimasi N, Bocciardi R, Di Battista E, Pramparo T, et al. Identification and molecular modelling of a novel familial mutation in the SRY gene implicated in the pure gonadal dysgenesis. Eur J Hum Genet 2007; 15: 76-80.
- 8. Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Arch Dis Child 2006; 91: 554-563.
- 9. Knower KC, Kelly S, Harley VR. Turning on the male - SRY, SOX9 and sex determination in mammals. Cytogenet Genome Res 2003; 101: 185-198.
- 10. Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, et al. The Human Gene Mutation Database: 2008 update. Genome Med 2009; 1: 13.
- 11. Shahid M, Dhillion VS, Jain N, Hedau S, Diwakar S, Sachdeva P, et al. Two new novel point mutations localized upstream and downstream of the HMG box region of the SRY gene in three Indian 46,XY females with sex reversal and gonadal tumour formation. Mol Hum Reprod 2004; 10: 521-526.
- 12. Marchina E, Gambera A, Spinelli E, Clerici P, Scagliola P, Sartori E, et al. Identification of a new mutation in the SRY gene in a 46,XY woman with Swyer syndrome. Fertil Steril 2009; 91: 932.
- 13. Assumpcao JG, Benedetti CE, Maciel-Guerra AT, Guerra G Jr, Baptista MT, Scolfaro MR, et al. Novel mutations affecting SRY DNA-binding activity: the HMG box N65H associated with 46,XY pure gonadal dysgenesis and the familial non-HMG box R30I associated with variable phenotypes. J Mol Med 2002; 80: 782-790.
- 14. Plaseska-Karanfilska D, Noveski P, Kuzevska K, Basheska N, Kocova M, Efremov GD. A new familial mutation (R133G) in the SRY gene. Clin Genet 2007; 71: 480-482.
- 15. Sambrook J, Russell DW. Molecular cloning: a laboratory manual 3rd edn. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001.
- 16. Schmitt-Ney M, Thiele H, Kaltwasser P, Bardoni B, Cisternino M, Scherer G. Two novel SRY missense mutations reducing DNA binding identified in XY females and their mosaic fathers. Am J Hum Genet 1995; 56: 862-869.
- 17. Hawkins JR, Taylor A, Goodfellow PN, Migeon CJ, Smith KD, Berkovitz GD. Evidence for increased prevalence of SRY mutations in XY females with complete rather than partial gonadal dysgenesis. Am J Hum Genet 1992; 51: 979-984.
- 18. Rimini R, Pontiggia A, Spada F, Ferrari S, Harley VR, Goodfellow PN, et al. Interaction of normal and mutant SRY proteins with DNA. Philos Trans R Soc Lond B Biol Sci 1995; 350: 215-220.
- 20. Harley VR, Clarkson MJ, Argentaro A. The molecular action and regulation of the testis-determining factors, SRY (sex-determining region on the Y chromosome) and SOX9 [SRY-related high-mobility group (HMG) box 9]. Endocr Rev 2003; 24: 466-487.
- 21. Murphy EC, Zhurkin VB, Louis JM, Cornilescu G, Clore GM. Structural basis for SRY-dependent 46-X,Y sex reversal: modulation of DNA bending by a naturally occurring point mutation. J Mol Biol 2001; 312: 481-499.
- 22. Shahid M, Dhillon VS, Hussain Z, Masa JF, Aslam M, Raish M, et al. Analysis of the SRY gene in two sex-reversed XY sisters identifies two new novel point mutations in the high mobility group box domain. Fertil Steril 2008; 90: 1199.e1-8.
Correspondence and Footnotes
Publication Dates
-
Publication in this collection
11 Nov 2011 -
Date of issue
Apr 2011
History
-
Accepted
31 Jan 2011 -
Received
07 Oct 2010