Abstract
Marfan syndrome (MFS) is an autosomal dominant disease of the connective tissue that affects the ocular, skeletal and cardiovascular systems, with a wide clinical variability. Although mutations in the FBN1 gene have been recognized as the cause of the disease, more recently other loci have been associated with MFS, indicating the genetic heterogeneity of this disease. We addressed the issue of genetic heterogeneity in MFS by performing linkage analysis of the FBN1 and TGFBR2 genes in 34 families (345 subjects) who met the clinical diagnostic criteria for the disease according to Ghent. Using a total of six microsatellite markers, we found that linkage with the FBN1 gene was observed or not excluded in 70.6% (24/34) of the families, and in 1 family the MFS phenotype segregated with the TGFBR2 gene. Moreover, in 4 families linkage with the FBN1 and TGFBR2 genes was excluded, and no mutations were identified in the coding region of TGFBR1, indicating the existence of other genes involved in MFS. Our results suggest that the genetic heterogeneity of MFS may be greater that previously reported.
Marfan syndrome; Fibrillin-1; TGF-β; Genetic heterogeneity
Abstract
Introduction
Material and Methods
Results
Discussion
References
Braz J Med Biol Res, August 2011, Volume 44(8) 793-800
Candidate gene linkage analysis indicates genetic heterogeneity in Marfan syndrome
L.V.S. Teixeira1, K.L. Mandelbaum2, L.V. Pereira1* and  Correspondence and Footnotes
Correspondence and Footnotes  A.B.A. Perez3*
A.B.A. Perez3*
1Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, 2Laboratório de Otorrinolaringologia/LIM32, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil
3Centro de Genética Médica, Departamento de Morfologia e Genética, Universidade Federal de São Paulo, São Paulo, SP, Brasil
 Correspondence and FootnotesCorrespondence and FootnotesCorrespondence and Footnotes
Correspondence and FootnotesCorrespondence and FootnotesCorrespondence and Footnotes
Marfan syndrome (MFS) is an autosomal dominant disease of the connective tissue that affects the ocular, skeletal and cardiovascular systems, with a wide clinical variability. Although mutations in the FBN1 gene have been recognized as the cause of the disease, more recently other loci have been associated with MFS, indicating the genetic heterogeneity of this disease. We addressed the issue of genetic heterogeneity in MFS by performing linkage analysis of the FBN1 and TGFBR2 genes in 34 families (345 subjects) who met the clinical diagnostic criteria for the disease according to Ghent. Using a total of six microsatellite markers, we found that linkage with the FBN1 gene was observed or not excluded in 70.6% (24/34) of the families, and in 1 family the MFS phenotype segregated with the TGFBR2 gene. Moreover, in 4 families linkage with the FBN1 and TGFBR2 genes was excluded, and no mutations were identified in the coding region of TGFBR1, indicating the existence of other genes involved in MFS. Our results suggest that the genetic heterogeneity of MFS may be greater that previously reported.
Key words: Marfan syndrome; Fibrillin-1; TGF-β; Genetic heterogeneity
Marfan syndrome (MFS; MIM 154700) is a relatively common autosomal dominant hereditary disorder of connective tissue (1:5,000-10,000 individuals) with prominent manifestations in the skeletal, ocular, and cardiovascular systems. Many affected individuals have a characteristic habitus with tall stature, long slender limbs (dolichostenomelia), arachnodactyly, scoliosis, and pectus excavatum or carinatum. Ectopia lentis affects up to 80% of individuals with MFS and is almost always bilateral (1-3). The leading cause of premature death in untreated individuals with MFS is acute aortic dissection, which follows a period of progressive dilatation of the ascending aorta (4-6).
The disease is caused by mutations in the FBN1 gene at 15q21.1, encoding the large cysteine-rich extracellular matrix glycoprotein fibrillin-1, the major component of microfibrils. FBN1 spans a 230-kb genomic region with 65 exons and about 600 different reported mutations spread throughout the gene, mostly specific to each affected family (7-13).
In 2003, an unexpected link between fibrillin-1 mutations and increased transforming growth factor-β (TGF-β) signaling was established in a mouse model for MFS, revealing a novel mechanism for the pathogenesis of the condition (14,15). Subsequently, it was found that the TGF-β receptor II gene (TGFBR2) was mutated in patients with MFS not linked to FBN1 (16). This provided the first genetic evidence of a direct link between abnormal TGF-β signaling and a human connective tissue disorder. Finally, it was proposed that mutations in the TGFBR1 or TGFBR2 gene lead to a different disease, Loeys-Dietz syndrome (LDS), with some clinical overlap with MFS (17). LDS is characterized by aortic dilatation, ocular hypertelorism, cleft lip/palate, arterial tourtoise, craniostenosis, and mental retardation. Nevertheless, there are reports of TGFBR1, TGFBR2, and FBN1 mutations in classical MFS, non-classical MFS, thoracic aortic aneurysms and dissections (TADD), and Shprintzen-Goldberg syndrome. In neonatal MFS, ectopia lentis and Weill-Marchesani syndrome, mutations only in FBN1 have been reported, whereas mutations associated with LDS have been found exclusively in the TGFBR1 and TGFBR2 genes (18,19).
At first, identification of mutations in the FBN1 gene appeared as the ultimate tool for confirmation of clinical diagnosis and risk assessment in MFS. As an alternative to the impractical direct identification of the disease-causing mutations in a gene as large as FBN1, in familial cases a successful approach to the molecular diagnosis is represented by linkage analysis using four intragenic microsatellite markers (20). Their segregation in families allows the identification of the disease haplotype.
In this study, we addressed the issue of genetic heterogeneity of MFS phenotypes by analyzing 34 families in a large cohort of Brazilian patients who met the clinical diagnostic criteria for the disease. We found 6 families in which linkage of the MFS phenotype with FBN1 was excluded, corroborating the hypothesis of genetic heterogeneity in this disease.
Subjects
Forty-seven MFS families (a total of 358 subjects) were referred to us by medical geneticists from the Medical Genetics Center (UNIFESP). All the probands met the diagnostic criteria for MFS (Ghent criteria) (6). The study was approved by the Institutional Ethics Committee of the Instituto de Biociências, Universidade de São Paulo, and all subjects gave written informed consent. Genomic DNA was extracted from leukocytes isolated from EDTA-anticoagulated whole blood (21).
Polymorphic markers
Four microsatellite markers, mts1, mts2, mts3, mts4, mapped to intron 1, 5, 28, and 43 of FBN1, respectively, were used for linkage analysis as previously described (20). The microsatellite markers used for TGFBR2 were D3S3727 (an intragenic TGFBR2 marker) and D3S3567. PCR primers and conditions were those described elsewhere (18). Fluorescently labeled PCR products were analyzed with the MegaBACE 1000 DNA Analysis system using the Genetic Profiler software, version 1.5 (Amersham Biosciences, Sweden).
Mutation analysis
The coding regions of the TGFBR1 gene were PCR amplified from genomic DNA, and PCR products were subjected to direct sequencing (18,22).
Lod score calculations
Lod score was calculated using the computer packages MLINK of the FASTLINK version 4.2 for two-point linkage analysis (23), and MERLIN for multipoint analysis (24). The disease was coded as fully penetrant, while the disease allele frequency was set at 0.0001. Meiotic recombination frequencies were considered to be equal for males and females. Allele frequencies for the microsatellite markers were calculated by the software based on data from each family. Maximum lod score for each family was calculated by simulation, considering that all meioses were informative. Families were scored according to segregation of markers and phenotypes as: linkage (lod score ≥3.0); linkage not excluded (-2 < lod score < 3.0), or linkage excluded (lod score < -2).
Statistical analyses
Statistical analysis was performed using the BioEstat software version 5.0. The yield of mutation detection in probands with involvement in each separate organ system was confirmed by the Pearson chi-square test, or the Fisher exact test for small samples. A P value of <0.05 was considered to be significant.
A total of 358 affected and unaffected individuals from 47 unrelated families were clinically assessed (). Although all probands met the Ghent diagnostic criteria for MFS, the clinical phenotype varied within and between families, as expected. The classic skeletal phenotype was observed in all 47 families, whereas in 67.6%, at least 1 individual had ectopia lentis, and in 82.3%, at least 1 individual showed dilatation of the aorta ().
Candidate gene linkage analysis indicates genetic heterogeneity in Marfan syndrome. L.V.S. Teixeira, K.L. Mandelbaum, L.V. Pereira and A.B.A. Perez. Braz J Med Biol Res 2011; 44: 793-800.
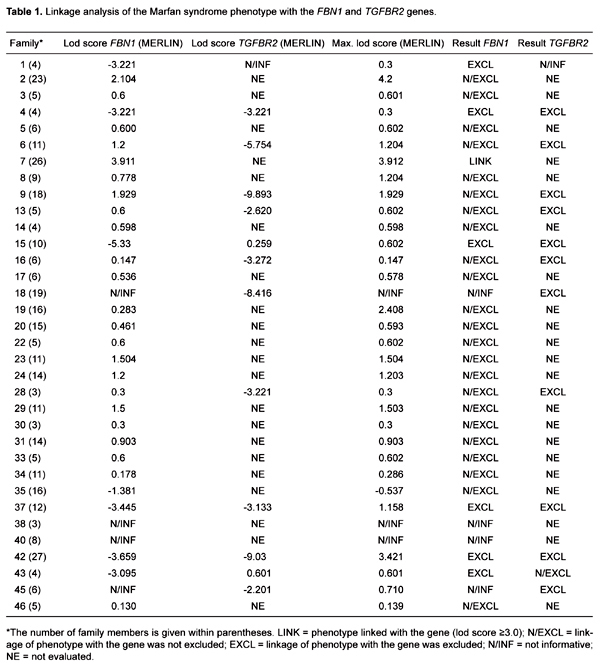
Thirty-four families presented enough individuals for linkage studies to be performed. Since most of the families were not large enough to obtain a lod score above 3.0, we compared the lod score obtained for each family with the theoretical maximum lod score possible for that family (Table 1). The microsatellite marker mts3 was not informative in many individuals, and was thus excluded from the analysis (data not shown). Initially we sought to identify those families where linkage to FBN1 could be excluded. We could not exclude linkage of the FBN1 haplotype with the MFS phenotype in 25 families (70.6%). A lod score greater than 3.0 was obtained for 1 of these families (family 7; Table 1). In the other families, the results of lod scores were below 3.0 due to the small number of individuals available for analysis. In 4 families (11.76%) the FBN1 gene markers were not informative, and thus the analysis was inconclusive. Finally, in 6 families (17.64%), we excluded segregation of FBN1 with the phenotype.
The 6 families excluded for linkage with FBN1 were studied with polymorphic markers linked to the TGFBR2 gene. In one family (family 43), linkage with the D3S3727 marker was not excluded, and one family was not informative for both TGFBR2 markers. However, in 4 of the 6 families (families 4, 15, 37, and 42; Table 1) we excluded segregation of TGFBR2 with the phenotype. Probands of these 4 families were screened for mutations in the coding region of TGFBR1, but no alteration was identified.
We compared the frequency of major manifestations in each of the three affected systems in the families whose phenotype segregated with FBN1 with those that did not segregate with FBN1 (Table 2). We found no significant difference in the frequency of cardiac and skeletal signs, but the major manifestations in the ocular system were significantly more frequent in families segregating with FBN1 than in those that did not segregate with this gene (Table 2).
Frequency of major signs in the three affected systems in families according to linkage with FBN1.
The discovery that mutations in gene FBN1 cause MFS has increased the chances of identifying the disease in atypical or oligosymptomatic individuals through molecular diagnosis (25). However, the more recent descriptions of mutations in other genes causing phenotypes overlapping with MFS have suggested the existence of genetic heterogeneity, or of a novel clinical entity, the LDS (18,22,25-27). The objective of the present study was to address the issue of genetic heterogeneity in MFS by characterizing a large cohort of patients with MFS phenotypes according to the gene linked to the phenotype, looking for clinical manifestations more frequent in the different groups.
We performed linkage analysis in 34 families with MFS phenotypes, which were evaluated clinically according to the criteria of Ghent. Linkage analysis performed with intragenic molecular markers in the FBN1 gene has been used by several investigators for the diagnosis of MFS (28-31). The effectiveness of using the analysis of haplotype segregation in families with oligosymptomatic individuals, or even with individuals clinically diagnosed with MFS, has been well demonstrated (20).
In most of the families analyzed, linkage of the MFS phenotype with the FBN1 gene was not excluded. However, we excluded linkage of the disease to this gene in 6 families whose affected individuals met the Ghent criteria for MFS, showing that either those diagnostic criteria are limited, or that the MFS phenotype can be caused by mutations in genes other than FBN1. We found a significant increase in the frequency of major ocular manifestations in families where linkage to FBN1 was not excluded than in those where linkage to this gene was excluded, where none of the patients had major ocular signs. Indeed, the lower frequency of ocular manifestations in patients with MFS-like phenotypes but without FBN1 mutations has been reported by others (12,13,19).
Among the 6 MFS families where linkage with FBN1 was excluded, we could not exclude linkage of the phenotype with TGFBR2 in one. However, the affected individuals did not meet the diagnostic criteria for LDS, corroborating the observations that mutations in the TGFBR2 gene can cause MFS (12,13,19). Finally, in 4 of those 6 families we excluded linkage of the MFS phenotype with TGFBR2, and found no mutations in the coding region of the TGFBR1 gene. Although one cannot rule out the existence of disease-causing mutations in promoter or intronic regions of TGFBR1, these results indicate the existence of additional genes that can lead to MFS.
In conclusion, the analysis of a large cohort of Brazilian families with MFS indicated that FBN1 was the gene most frequently involved in the disease, mostly when there were major ocular findings. However, this study also identified families where the MFS phenotype did not segregate with FBN1, revealing genetic heterogeneity in this disease. Particularly, the 4 families that did not have mutations in either the TGFBR1 or TGFBR2 gene, although small, represented an opportunity to search for other genes involved in the MFS phenotypes. The identification of these genes should lead to a better understanding of the molecular mechanisms involved in the normal and pathogenic physiology of the ocular, skeletal and cardiovascular systems.
1. Pyeritz RE. The Marfan syndrome. Am Fam Physician 1986; 34: 83-94.
2. Pyeritz RE. Marfan syndrome. In: Emery AEH, Rimoin DL (Editors), Principles and practice of medical genetics. 2nd edn. New York: Churchill Livingstone; 1990. p 1047-1063.
3. Pyeritz RE, Francke U. The Second International Symposium on the Marfan Syndrome. Am J Med Genet 1993; 47: 127-135.
4. Summers KM, West JA, Peterson MM, Stark D, McGill JJ, West MJ. Challenges in the diagnosis of Marfan syndrome. Med J Aust 2006; 184: 627-631.
5. Beighton P. McKusick’s heritable disorders connective tissue. 5th edn. St. Louis: Mosby; 1993.
6. De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996; 62: 417-426.
7. Kainulainen K, Savolainen A, Palotie A, Kaitila I, Rosenbloom J, Peltonen L. Marfan syndrome: exclusion of genetic linkage to five genes coding for connective tissue components in the long arm of chromosome 2. Hum Genet 1990; 84: 233-236.
8. Dietz HC, Cutting GR, Pyeritz RE. Defects in the fibrillin gene cause the Marfan syndrome; linkage evidence and identification of a missense mutation. Nature 1991; 352: 37-39.
9. Tsipouras P, Sarfarazi M, Devi A, Weiffenbach B, Boxer M. Marfan syndrome is closely linked to a marker on chromosome 15q1.5----q2.1. Proc Natl Acad Sci U S A 1991; 88: 4486-4488.
10. Pereira L, D’Alessio M, Ramirez F, Lynch JR, Sykes B, Pangilinan T, et al. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum Mol Genet 1993; 2: 961-968.
11. Perez ABA, Pereira LVP, Zatz M, Brunoni D, Passos-Bueno MR. A genetic, clinical and molecular approach in Brazilian families with Marfan syndrome. Hum Mutat 1998; 13: 84-87.
12. Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007; 81: 454-466.
13. Faivre L, Collod-Beroud G, Child A, Callewaert B, Loeys BL, Binquet C, et al. Contribution of molecular analyses in diagnosing Marfan syndrome and type I fibrillinopathies: an international study of 1009 probands. J Med Genet 2008; 45: 384-390.
14. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003; 116: 217-224.
15. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003; 33: 407-411.
16. Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet 2006; 43: 769-787.
17. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275-281.
18. Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 2004; 36: 855-860.
19. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476-485.
20. Pereira L, Levran O, Ramirez F, Lynch JR, Sykes B, Pyeritz RE, et al. A molecular approach to the stratification of cardiovascular risk in families with Marfan’s syndrome. N Engl J Med 1994; 331: 148-153.
21. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
22. Matyas G, Arnold E, Carrel T, Baumgartner D, Boileau C, Berger W, et al. Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum Mutat 2006; 27: 760-769.
23. Schaffer AA. Faster linkage analysis computations for pedigrees with loops or unused alleles. Hum Hered 1996; 46: 226-235.
24. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97-101.
25. Boileau C, Alexandre JA, Hariti G, Babron MC, Coulon M, Salvat C, et al. Evidence for genetic heterogeneity in Marfan syndrome. Cytogenet Cell Genet 1991; 58: 1991-1996.
26. Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, et al. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat 2006; 27: 770-777.
27. Sakai LY, Ikegawa S, Ito E, Numabe H, Watanabe Y, Mikami H, et al. Cohompreensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am J Med Genet 2006; 140 A: 1719-1725.
28. Mottes M, Mirandola S, Rigatelli F, Zolezzi F, Lisi V, Gordon D, et al. Allelic frequencies of FBN1 gene polymorphisms and genetic analysis of Italian families with Marfan syndrome. Hum Hered 2000; 50: 175-179.
29. Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet 2003; 12: 2269-2276.
30. Lee NC, Hwang B, Chen CH, Niu DM. Intrafamilial phenotype variation in Marfan syndrome ascertained by intragenic linkage analysis. J Formos Med Assoc 2005; 104: 964-967.
31. Spits C, De Rycke M, Verpoest W, Lissens W, Van Steirteghem A, Liebaers I, et al. Preimplantation genetic diagnosis for Marfan syndrome. Fertil Steril 2006; 86: 310-320.

Clinical and genetic assessment of Marfan syndrome patients in 47 families.
Address for correspondence: A.B.A. Perez, Centro de Genética Médica, Departamento de Morfologia e Genética, UNIFESP, Rua Coronel Lisboa, 966, 04020-041 São Paulo, SP, Brasil. E-mail: anabia.morf@epm.br
*These authors contributed equally to this study. Received January 27, 2011. Accepted July 13, 2011. Available online July 29, 2011. Published August 19, 2011.
The Brazilian Journal of Medical and Biological Research is partially financed by






All the contents of this journal, except where otherwise noted, is licensed under a Creative Commons Attribution License
- 1. Pyeritz RE. The Marfan syndrome. Am Fam Physician 1986; 34: 83-94.
- 2. Pyeritz RE. Marfan syndrome. In: Emery AEH, Rimoin DL (Editors), Principles and practice of medical genetics 2nd edn. New York: Churchill Livingstone; 1990. p 1047-1063.
- 3. Pyeritz RE, Francke U. The Second International Symposium on the Marfan Syndrome. Am J Med Genet 1993; 47: 127-135.
- 4. Summers KM, West JA, Peterson MM, Stark D, McGill JJ, West MJ. Challenges in the diagnosis of Marfan syndrome. Med J Aust 2006; 184: 627-631.
- 6. De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996; 62: 417-426.
- 7. Kainulainen K, Savolainen A, Palotie A, Kaitila I, Rosenbloom J, Peltonen L. Marfan syndrome: exclusion of genetic linkage to five genes coding for connective tissue components in the long arm of chromosome 2. Hum Genet 1990; 84: 233-236.
- 8. Dietz HC, Cutting GR, Pyeritz RE. Defects in the fibrillin gene cause the Marfan syndrome; linkage evidence and identification of a missense mutation. Nature 1991; 352: 37-39.
- 9. Tsipouras P, Sarfarazi M, Devi A, Weiffenbach B, Boxer M. Marfan syndrome is closely linked to a marker on chromosome 15q1.5----q2.1. Proc Natl Acad Sci U S A 1991; 88: 4486-4488.
- 11. Perez ABA, Pereira LVP, Zatz M, Brunoni D, Passos-Bueno MR. A genetic, clinical and molecular approach in Brazilian families with Marfan syndrome. Hum Mutat 1998; 13: 84-87.
- 12. Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007; 81: 454-466.
- 13. Faivre L, Collod-Beroud G, Child A, Callewaert B, Loeys BL, Binquet C, et al. Contribution of molecular analyses in diagnosing Marfan syndrome and type I fibrillinopathies: an international study of 1009 probands. J Med Genet 2008; 45: 384-390.
- 14. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003; 116: 217-224.
- 15. Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003; 33: 407-411.
- 16. Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet 2006; 43: 769-787.
- 17. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275-281.
- 18. Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 2004; 36: 855-860.
- 19. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476-485.
- 21. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
- 22. Matyas G, Arnold E, Carrel T, Baumgartner D, Boileau C, Berger W, et al. Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum Mutat 2006; 27: 760-769.
- 23. Schaffer AA. Faster linkage analysis computations for pedigrees with loops or unused alleles. Hum Hered 1996; 46: 226-235.
- 24. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97-101.
- 25. Boileau C, Alexandre JA, Hariti G, Babron MC, Coulon M, Salvat C, et al. Evidence for genetic heterogeneity in Marfan syndrome. Cytogenet Cell Genet 1991; 58: 1991-1996.
- 26. Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, et al. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat 2006; 27: 770-777.
- 27. Sakai LY, Ikegawa S, Ito E, Numabe H, Watanabe Y, Mikami H, et al. Cohompreensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am J Med Genet 2006; 140 A: 1719-1725.
- 28. Mottes M, Mirandola S, Rigatelli F, Zolezzi F, Lisi V, Gordon D, et al. Allelic frequencies of FBN1 gene polymorphisms and genetic analysis of Italian families with Marfan syndrome. Hum Hered 2000; 50: 175-179.
- 29. Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet 2003; 12: 2269-2276.
- 30. Lee NC, Hwang B, Chen CH, Niu DM. Intrafamilial phenotype variation in Marfan syndrome ascertained by intragenic linkage analysis. J Formos Med Assoc 2005; 104: 964-967.
- 31. Spits C, De Rycke M, Verpoest W, Lissens W, Van Steirteghem A, Liebaers I, et al. Preimplantation genetic diagnosis for Marfan syndrome. Fertil Steril 2006; 86: 310-320.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
27 Sept 2011 -
Date of issue
Aug 2011
History
-
Received
27 Jan 2011 -
Accepted
13 July 2011