Abstract
The transient receptor potential channels family (TRP channels) is a relatively new group of cation channels that modulate a large range of physiological mechanisms. In the nervous system, the functions of TRP channels have been associated with thermosensation, pain transduction, neurotransmitter release, and redox signaling, among others. However, they have also been extensively correlated with the pathogenesis of several innate and acquired diseases. On the other hand, the omega-3 polyunsaturated fatty acids (n-3 fatty acids) have also been associated with several processes that seem to counterbalance or to contribute to the function of several TRPs. In this short review, we discuss some of the remarkable new findings in this field. We also review the possible roles played by n-3 fatty acids in cell signaling that can both control or be controlled by TRP channels in neurodegenerative processes, as well as both the direct and indirect actions of n-3 fatty acids on TRP channels.
TRP channels; Calcium; Omega-3 fatty acids; Reactive oxygen species; G-protein coupled receptors; Neurodegeneration
Abstract
Introduction
References
Acknowledgments
Braz J Med Biol Res, November 2011, Volume 44(11) 1088-1096
TRP channels, omega-3 fatty acids, and oxidative stress in neurodegeneration: from the cell membrane to intracellular cross-links
 Correspondence and Footnotes
Correspondence and Footnotes
 M. Leonelli, M.F.R. Graciano and L.R.G. Britto
M. Leonelli, M.F.R. Graciano and L.R.G. Britto
Departamento de Fisiologia e Biofísica, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, SP, Brasil
 Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
The transient receptor potential channels family (TRP channels) is a relatively new group of cation channels that modulate a large range of physiological mechanisms. In the nervous system, the functions of TRP channels have been associated with thermosensation, pain transduction, neurotransmitter release, and redox signaling, among others. However, they have also been extensively correlated with the pathogenesis of several innate and acquired diseases. On the other hand, the omega-3 polyunsaturated fatty acids (n-3 fatty acids) have also been associated with several processes that seem to counterbalance or to contribute to the function of several TRPs. In this short review, we discuss some of the remarkable new findings in this field. We also review the possible roles played by n-3 fatty acids in cell signaling that can both control or be controlled by TRP channels in neurodegenerative processes, as well as both the direct and indirect actions of n-3 fatty acids on TRP channels.
Key words: TRP channels; Calcium; Omega-3 fatty acids; Reactive oxygen species; G-protein coupled receptors; Neurodegeneration
Ca2+ signaling is a ubiquitous mechanism in the control of cell function. Many cellular processes depend on Ca2+ levels or on Ca2+ transients for the control of enzyme activation, gene transcription, membrane potential, and cytoskeleton remodeling, among other processes. These processes, in turn, are responsible for neuronal activation and neurotransmitter release, control of cell proliferation and cell death, hormonal balance, and muscle contraction (1).
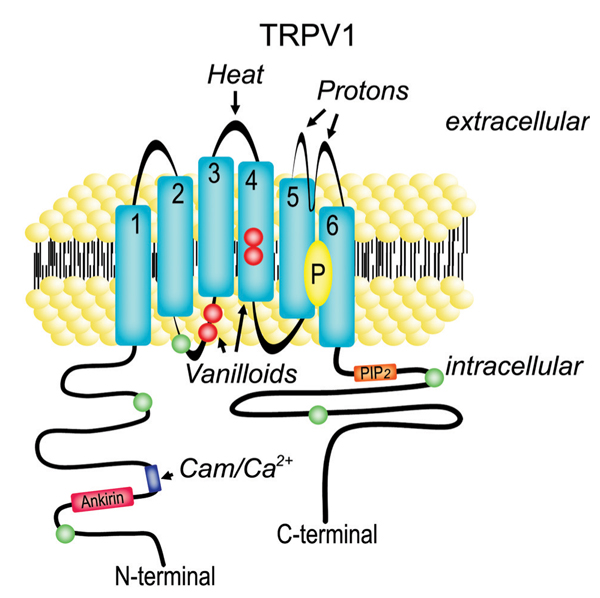
The transient receptor potential channels (TRP channels) comprise a relatively recently identified group of cationic permeable channels, which have distinct Ca2+ selectivity (2). In mammals, the 28 members so far identified of this family are divided into 6 subgroups in terms of protein homology: canonical, TRPC channels; vanilloid, TRPV channels; melastatin, TRPM channels; ankyrin, TRPA channels; mucolipins, TRPML channels, and polycystins, TRPP channels (2,3). TRP channels are 6 transmembrane-spanning proteins with both amino and carboxy tails located on the intracellular side of the membrane. The basic structure of a prototypic TRP channel, the TRPV1 channel, is depicted in Figure 1. These channels can be differentially modulated by a large number of stimuli, including downstream effectors of G-protein-coupled receptors, calmodulin, temperature, protons, hydrogen peroxide, depletion of Ca2+ stores, cations such as Ca2+ and Mg2+, and ligands, which are found in several ingredients commonly used in worldwide cuisines, such as garlic, vanilla, red peppers, and cinnamon, and still other substances that are used in medicine, such as camphor and eugenol (4,5). However, just a few direct endogenous modulators of TRP have been recognized so far, and they include some arachidonic acid-derived lipids, such as the biogenic amines anandamide and N-arachidonoyldopamine (NADA), and the lipoxygenase products 12(S) and 15(S) HPETE and leukotriene B4 (6). The function of TRP channels can also be controlled by several protein kinases, which in turn modulate the sensitivity and the intracellular traffic of these channels (4,7).
Ca2+ flux through TRP channels located in the plasma membrane and in the membranes of intracellular organelles of excitable and non-excitable cells can promote changes in intracellular-free Ca2+ concentrations and in the membrane potential, which can modulate the driving force for other ions and for Ca2+ itself (8). In the central nervous system, TRP channels are involved in many physiological processes, including the development of neuronal tissue, neuronal sprouting, cell communication, microglial activation, hydroelectrolytic balance, control of body temperature, and sensory signaling (7).
In the central nervous system, TRP channels can contribute to excitatory neurotransmitter release (9), redox signaling (10-15), and modulation of mitochondrial Ca2+ levels (16). Normally, these functions of TRP channels can aid normal cell signaling, but their excessive mobilization of intracellular pathways can exacerbate the TRP-linked cascade and may eventually cause the loss of Ca2+ homeostasis, triggering cell suffering and death (12,17,18). It is noteworthy that, to date, at least 13 human diseases have been linked to TRP mutations (19), such as mucolipidosis type 4, caused by specific mutations in TRPML1 channels (20). Several neuronal deficits and behavioral impairment have also been observed in different TRP knockout models (21,22). Additionally, TRP channel function has been associated with the onset and the progression of several neurodegenerative diseases, such as Alzheimer (23), Parkinson (24,25), stroke and hypoxia (11,12), among others. The main mechanisms that underlie the roles played by TRP channels in disease were recently summarized by Nilius (26).
In this review, we intend to summarize some recent intriguing findings that correlate TRP function with some polyunsaturated fatty acids (PUFAs), especially the omega-3 (n-3) fatty acids. The roles played by n-3 fatty acids in cell signaling pathways that can both control or be controlled by TRP channels in neurodegenerative processes will be emphasized. Some protein-lipid interactions that occur on the cell membrane and other direct actions of n-3 fatty acids on TRP function and possible cross-talks that can occur between their downstream pathways will be also discussed.
TRP channels are involved in excitotoxicity and oxidant-induced cell toxicity
A large number of TRP channels permit the influx of large amounts of Ca2+ (8) and might be involved in excitotoxicity and oxidative damage in neuronal tissue. Both processes have been extensively correlated with the emergence and progression of a large number of neurodegenerative diseases that share Ca2+ imbalance and consequent oxidative-mediated neuronal injury as triggering and maintaining processes (27).
Excitotoxicity is known to cause cell death mediated by profuse cytoplasmic Ca2+ entry through N-methyl-D-aspartic acid (NMDA) and non-NMDA glutamate receptors, as well as through L-type voltage-gated channels, leading to consequent protease activation and reactive oxygen and nitrogen species (ROS and RNS, respectively) production (28). Anti-excitotoxic therapy is a strategy used for the treatment of stroke in clinical practice, which consists of the antagonism of such channels, but which cannot completely abrogate neuronal death (29). At the onset of hypoxia and glucose deprivation, disruption of mitochondrial function and depletion of ATP reduces the synaptic capability to control both neurotransmitter release and its clearance from the synaptic cleft, causing massive activation of glutamate receptors at first, and therefore desensitizing such receptors (28). The main effects of anti-excitotoxic therapy is observed when treatment is applied during the first minutes after hypoxia, strongly suggesting that glutamate excitotoxicity plays a role in neuronal cell death only in the first minutes after hypoxia and glucose deprivation (29).
The second phase of cell death that occurs in hypoxia and glucose deprivation seems not to depend on the activation of amino acid receptors (28), possibly indicating that other ionic channels might play a role in this process. It was recently shown that Ca2+ conductance through TRPM7 is an important requirement for triggering neuronal death in oxygen and glucose deprivation models (12). In fact, both TRPM2 and TRPM7 channels seem to be involved in neuronal cell death induced by oxidative stress. These proteins form Ca2+-permeable channels that are gated by ROS and RNS. TRPM7 blockade reduces cell death in ischemic models. Ca2+ influx through those channels is also responsible for ROS and RNS production, which positively regulate TRPM7 function. These results are particularly relevant because they indicate that ROS and RNS production induced by TRPM7 currents may cause a positive feedback loop responsible for the exacerbation of cell death in prolonged hypoxia and glucose deprivation (12).
Interestingly, Mg2+ homeostasis may also contribute to ROS-mediated TRP signaling. Free Mg2+ or Mg2+-ATP is able to inhibit TRPM7 channel ion flux (30). It is believed that TRPM7 channels are a main route for Mg2+ influx in the central nervous system (31), whereas Mg2+/ATP-dependent antiporters mediate the extrusion of Mg2+ in exchange for other ions. When ATP is low, the intracellular concentration of free Mg2+ is assumed to rise (32,33). The rise of intracellular Mg2+ could thus inhibit TRPM7. However, it is not yet known which signaling pathway is most relevant for TRPM7 function in vivo, whether that mediated by Mg2+ or the one mediated by ROS/RNS. It is possible that normal oxygen and glucose level restoration (reperfusion) would induce the normal functioning of Mg2+/ATP-dependent pumps, thus reducing the inhibition of TRPM7 channels caused by Mg2+. Additionally, it is well documented that reperfusion allows the increase of ROS and RNS levels and cell damage mediated by such species, which is mainly due to severe reduction of antioxidant defenses (34), and a vicious cycle could be triggered by the activation of TRPM7 and consequent ROS/RNS production due to Ca2+ overload. Strengthening this idea, TRPM7 suppression with viral vectors bearing shRNA protected hippocampal CA1 neurons and preserved both cell morphology and function against ischemia-induced cell death (11). Therefore, it seems clear that TRPM7 channels are involved in ROS-mediated neuronal death.
TRPM2 channel function is also controlled by oxidants. H2O2 at micromolar concentrations that do not cause cell death directly, and the superoxide donor dithionite caused Ca2+ influx through TRPM2 channels in HEK cells. The reduction of both glutathione and thioredoxin function and the incubation of the nitric oxide (NO) donor SNAP potentiated the effects of H2O2 on Ca2+ influx. Moreover, H2O2 reduced cell viability in a dose-dependent manner (35). In neurons, micromolar H2O2 also activated TRPM2, leading to Ca2+ overload and cell death, and down-regulation of TRPM2 with siRNA decreased both Ca2+ influx and cell death elicited by H2O2 (36). Interestingly, H2O2 can also elicit TRPM2-mediated Ca2+ currents in microglial cells, and previous LPS treatment largely potentiated this effect (37), indicating that these receptors might be secondary modulators of microglial function. TRPM2 currents are also triggered by ADP-ribose. Oxidative and nitrosative stress can enhance ADP-ribose production by the nucleus and by mitochondrial metabolism (38). In the former case, oxidants can cause DNA damage, and the DNA repair machinery generates ADP-ribose as a by-product. It was recently demonstrated that H2O2 and the nitrosothiol donor N-methyl-N"-nitro-N-nitrosoguanidine enhanced ADP-ribose production, which in turn also caused TRPM2 Ca2+ influx (10). Taken together, these results indicate that TRPM2 might be a cell sensor for redox status in the normal and diseased neuronal tissue.
The function of other TRP channels such as TRPC3 and TRPC4 can also be triggered by oxidative stress (14,15). The mechanisms found so far to explain the sensitivity of TRPC channels to oxidants suggest that the final routes that culminate in TRPC activation are the activation of phospholipase C (PLC) and of tyrosine kinase receptors (RTKs). PLC and its downstream effectors can positively modulate the function of several TRPCs (5). Pharmacological inhibition of PLC prevented TRPC3-mediated cationic flux triggered by oxidants (15). On the other hand, expression, down-regulation and functional disruption of Src kinases also reduce TRPC3 activation. Given that Src kinases can be activated in oxidative conditions, it is at least suggestive that RTK signaling could have a role in oxidant-induced TRPC3 function.
Ca2+-mediated NO production can also ensue as a consequence of TRP activation. Some TRP channels can also control or be controlled by NO or NO-dependent signaling. NO can both mediate the nitrosylation of specific cysteine residues (S-nitrosylation) (39) or, after its reaction with other ROS, it can increase the generation of other oxidants involved in nitration (40). Our group has studied the participation of TRPV1 channels and correlated receptors in retinal development and neuronal degeneration that occur after optic nerve damage (13,41-43). It seems that signaling through TRPV1 channels is partially responsible for excessive NO production and consequent protein nitration in both retinal neuronal and glial cells (13). Although TRPV1 channels are not directly involved in acute cell death after optic nerve axotomy, our results indicate that their function collaborates with gliosis of Müller cells, which seems to occur as a result of increased protein nitration (13). Another mechanism that might be involved is that the direct activation of TRPV1 receptors could trigger glutamate release (9,16,44), which in turn could lead to excitotoxicity.
More research is clearly needed to elucidate the possible participation of these channels in processes in which signaling mediated by ROS and RNS is associated with neuronal cell death in the brain, such as stroke, epilepsy, and Alzheimer"s disease, among others. The development of selective drugs to modulate these channels will be certainly of great value for future research and for clinical trials.
Direct modulation of TRP channels by n-3 fatty acids
Possible direct and indirect interactions between n-3 fatty acids and TRP channels are summarized in Figure 2. n-3 fatty acids and their derivatives can control neuronal excitability through the modulation of several ionic channels. In the last few years, several lines of evidence have indicated that TRP channel function can be modulated both directly and indirectly by n-3 fatty acids. One of the most important findings in the field was reported by Ahern et al. (45). It was demonstrated that the n-3 fatty acids docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) at physiological concentrations have the ability to evoke small TRPV1 currents, which seems to be dependent on the previous sensitization of the channel by protein kinase C (PKC). DHA also reduced the voltage dependence for the activation of TRPV1, which could indicate that DHA can turn TRPV1 more prone to open at physiological voltages. On the other hand, these investigators also found that both DHA and EPA can displace [3H]-resiniferatoxin binding, while DHA, at concentrations that displace almost 100% of [3H]-resiniferatoxin binding, induced currents that can be considered small when compared to those elicited by the vanilloid capsaicin. In fact, α-linolenic acid, EPA, arachidonic acid, and γ-linolenic acid also reduced the current evoked by several vanilloids (45). These results suggest that these n-3 fatty acids can modulate TRPV1 directly at the vanilloid binding site, or that they could allosterically interact with the receptor, thus inducing changes in the channel structure that both avoid [3H]-resiniferatoxin binding and cause conformational changes responsible for channel opening. This idea is supported by the recent finding that DHA and other amphiphiles can increase membrane elasticity and can therefore modulate the function of membrane-embedded channels (46), which supports the general idea that the cellular lipid bilayer can also modulate protein function allosterically, including some ionic channels (47).
These results show clearly that TRPV1 currents are modulated by several PUFAs, which seem to reduce the ability of the vanilloid agonists to induce a large cationic influx. Supporting this idea, it has been demonstrated that α-linolenic acid and other n-3 fatty acids also inhibited the cold-sensitive TRPM8 and TRPV1 channels (48,49). Whether these effects are due to the action of these PUFAs on the agonist binding site or are due to conformational changes caused by different TRP protein interactions with the lipid bilayer still requires further experimental efforts.
The ability of n-3 PUFAs to reduce TRPV1 activation is extremely exciting, since it adds another type of interaction between TRPs and n-3 fatty acids different from the classical indirect relationship that is known to occur between n-3 fatty acids and n-6 fatty acids, arachidonic acid derivatives, which include some TRPV1 agonists. Several TRPV1 ligands are lipoxygenase derivatives of arachidonic acid (6). It seems that n-3 fatty acids are also able to compete with arachidonic acid as substrates for lipoxygenases and cyclooxygenases, possibly being implicated in the reduction of the final production of inflammatory mediators that could activate the TRPV1 channel (50). Despite the fact that these results are confined to TRPV1 channels, they may shed a new light on the research on the physiology of other TRP channels. Several other TRP channels such as TRPV3, TRPV4, and TRPA1 can be modulated by arachidonic acid and its derivatives (51-53), and the availability and disposal of arachidonic acid is essential for the production of such compounds (54). n-3 fatty acids can also compete with other fatty acids, including arachidonic acid, both reducing the precursors of inflammatory mediators and forming diverse diacylglycerol (DAG) compounds (55), which could reduce the activation of several protein kinases that differentially modulate the function of multiple TRP channels (7).
Indirect modulation of TRP channels by n-3 fatty acids
Epidemiological studies and animal models have demonstrated that PUFAs are crucial for the normal function of the brain. In aged rats, spatial memory and hippocampal long-term potentiation can be improved with dietary supplementation of arachidonic acid and the memory function of human subjects is significantly improved with PUFA supplementation (56-58). DHA is also essential for neural development and brain function (59), and is responsible for the antiapoptotic effect on neuronal death and the prevention of ischemic brain damage (60,61). In this regard, the activation of PUFA-sensitive receptors might have an important participation in these processes.
G-protein-coupled receptors (GPCRs), members of the large family of the seven transmembrane domain receptors, are known to play physiological roles in response to free fatty acids. GPR40 is a member of the subfamily of homologous GPCRs that include GPR41 and GPR43. GPR40 was reported to be preferentially expressed in human and rodent pancreas and in human brain, and the ligands of GPR40 are medium- and long-chain fatty acids, such as oleic, arachidonic and docosahexaenoic acids (62,63). Fatty acids binding to GPR40 activate a heterotrimeric G-protein containing the α subunit of the Gq protein, which is known to stimulate PLC activity. This enzyme converts phosphatidylinositol 3,5-bisphosphate to DAG and inositol triphosphate (IP3). DAG is a well-known PKC activator and IP3 participates in endoplasmic reticulum Ca2+ extrusion. PKC phosphorylates a large number of TRP channels at multiple sites, and several TRP channels can be sensitized by PKC, such as TRPV1, TRPV4 and TRPC1, whereas almost all other TRPC channels, in addition to TRPM4 and TRPM8, appear to be desensitized by PKC phosphorylation (64,65). Furthermore, DAG can modulate the gating of a large number of TRPCs, whereas IP3-mediated intracellular Ca2+ store depletion might activate a few TRPs (66).
The function of GPR40 in the nervous system is unknown. GPR40 is expressed in neurons of the cerebral cortex, hippocampus, amygdala, hypothalamus, cerebellum, substantia nigra, pons, medulla oblongata, spinal cord, and the pituitary gland. Furthermore, GPR40 was detected in astrocytes of the cerebral white matter, the molecular layer and multiform layer of the cerebral cortex, and in astrocytes within the progenitor cell niche, such as the subventricular zone (SVZ) along the anterior horn of the lateral ventricle, and the subgranular zone (SGZ) of the hippocampal dentate gyrus of adult monkeys (67). Endothelial cells of the SGZ express GPR40 (68), which could represent an important element of the angiogenic niche, where recruitment and subsequent remodeling are associated with neurogenesis. The ubiquitous distribution of this receptor in the primate brain suggests that fatty acids might act as extracellular signaling molecules to regulate neuronal function.
Increased expression of GPR40 in the SGZ niche has been demonstrated in the second week after global cerebral ischemia in monkeys (68). It has been speculated that the postischemic increase of GPR40 in endothelial cells and newborn astrocytes may be able to enhance local DHA concentration, since only astrocytes can synthesize DHA in the nervous system (69). Also, DHA may function as an extracellular signaling molecule leading to the generation and differentiation of neural progenitors and newborn neurons through the up-regulation and activation of GPR40 (68). In this process, fatty acid-binding proteins might participate in the maintenance of appropriate levels of PUFAs in the neurogenic niche, which are required for the postischemic neurogenesis, contributing to the proliferation of neural progenitor cells (70).
DHA-induced neuronal differentiation, neurite growth and branching of adult rat stem cells are all mediated by a mechanism that involves GPR40 signaling in rat neural stem cells (NSC) transfected with the GPR40 gene (71). Ca2+ mobilization induced by DHA in GPR40-expressing NSC was completely blocked by the inhibition of the IP3 receptor, an important element activated by GPR40 signaling (71).
It is extremely interesting to note that several Gq- and DAG-sensitive TRP channels such as TRPCs (5) have also been related to the extension of neurites and growth of neuronal cones (4). However, it still remains completely obscure if GPR40 receptors can modulate the function of TRP channels. Despite the recent cloning of GPR40 and the fact that available data in the literature on brain GPR40, although important, are still limited, it seems at least strongly suggestive that some cross-links might occur. Both GPR40 receptors and a large number of TRP channels are widely expressed in the central nervous system, and several subproducts of GPR40 activation are well-known TRP activators. The next question that could be asked to address this issue is where and how those signaling systems could share their function in neuronal tissue.
Recent findings also indicate that TRP channel function can be modulated by resolvins. Resolvins from the families "E" and "D" are oxidized products from the n-3 fatty acids EPA and DHA, respectively (72). Resolvin binding on GPCRs seems to be part of the mechanism that underlies their action. The function of resolvins has been extensively associated with cellular protection and cessation of the inflammatory signaling (72). Resolvin D1 reduced the activation of the channels TRPA1, TRPV3 and TRPV4 by specific agonists. However, the most exciting finding was that the blockade of specific GPCR cascades did not disrupt the attenuation of TRP channel activity, thus indicating that resolvin D1 may bind and block these TRP channels directly (73). On the other hand, it was shown that resolvin E1 reduced TRPV1-mediated glutamate release through its action on the Gαi-protein-coupled receptor ChemR23 (74). These results are extremely challenging, given that resolvins are lipoxygenase products as much as several endogenous ligands of TRPV1 receptors (6,72).
Since preferential metabolization of n-3 fatty acids or arachidonic acid by lipoxygenase seems to be dependent on diverse factors, such as previous acetylation of cyclooxygenase 2 by aspirin or by substrate disposal (50,72), any therapeutic attempt must consider that new drugs that may alter substrate availability, or even classic non-steroidal anti-inflammatories, might modulate directly or indirectly the signaling mediated by n-3 fatty acids in TRP channels.
TRPs can also be good, and n-3 fatty acids can harm
Existing evidence indicates that no general rule can be applied to the function of both n-3 fatty acids and TRP channels in the brain. The function of several TRP channels is extremely important for normal brain development (75) and neuronal survival in the adult. As an example, the function of TRPC1 channels has been implicated in the survival of SH-SY5Y human neuroblastoma cells treated with MPP+ (24), a widely used neurotoxin employed in animal models of Parkinson"s disease. TRPC1 receptors are highly expressed in the substantia nigra (76), and these results clearly show that the modulation of Ca2+ influx mediated by those channels might be an interesting pharmacological tool for future studies on Parkinson"s disease.
On the other hand, increased production of oxidant and oxidized products by n-3 fatty acids has been extensively demonstrated in tumors and tumoral cell lines (for a review, see Ref. 77). n-3 fatty acids and other PUFAs can alter mitochondrial metabolism, resulting in increased ROS production and mitochondrial respiratory chain impairment, which can ultimately lead to ATP depletion and ionic imbalance (77). In this scenario, cells might have no time to adapt to the new Ca2+ levels (78), and cell death may eventually occur. Furthermore, DHA-enriched diets were reported to increase the enzymatic activity of brain NO synthase (79), which might contribute to TRP channel opening. As mentioned before, NO can also participate in the process of S-nitrosylation, which is a physiological and reversible process controlled by NO levels and by the cellular redox status, thus enabling NO to modulate the function of many proteins (39). S-nitrosylation has been reported to modulate the function of several TRP channels, and it seems that nitrosylation increases the sensitivity of these channels (80). These few examples clearly show that the intermodulation of TRP channels and n-3 fatty acids is very far from any generalization, and a particular analysis concerning membrane interactions, cellular particularities, microenvironment, and the types of TRP channels and PUFAs, as well as other neurotransmitters and interacting pathways, must be cautiously considered before any analysis.
Conclusions
Considering the results obtained thus far, it seems that n-3 fatty acids tend to be neuroprotective, but it is impossible to assert that they have only a single and general implication in TRP function in the central nervous system. Instead, the above data indicate that n-3 fatty acids can modulate TRP at multiple levels. If, on the one hand, TRP function can be elicited by n-3 fatty acids, on the other hand, some reports indicate that the evoked currents are apparently smaller than those observed with other agonists. This indicates that n-3 fatty acids may attenuate large disparities in Ca2+ flow (i.e., no flow versus large flow), which may essentially be at the root of Ca2+ excitotoxicity. Furthermore, microenvironmental factors must be taken into account when dealing with the possible therapeutics of n-3 fatty acids applied to TRP-involved dysfunctions, given that neuronal cell populations of a given area might respond differently when facing similar drugs or when exposed to a particular lipid profile. It should also be remembered that tumors and tumoral cell lines, due to several factors including their large energy and protein synthesis demand, are expected to behave quite differently when compared to normal cells. In conclusion, the relationship of TRP channels and n-3 fatty acids is still far from being completely elucidated, although the few examples shown here are extremely encouraging for future research on the possible interactions between TRP channels and n-3 fatty acids under both physiological and pathological conditions.
Structure of the prototypical transient receptor potential (TRP) channel, TRPV1. The TRPV1 channel is a six transmembrane-spanning protein, and the pore (P, yellow) is formed by the fifth and sixth transmembrane domains. It has three ankirin repeats (pink box) and a Ca2+/calmodulin-interacting site (dark blue box) in the N-terminal ending. The C-terminal ending has an interacting site for phosphatidylinositol 4,5-bisphosphate (PIP2; orange box). Several residues (green circles) can be phosphorylated by both the protein kinases C and A (PKC and PKA, respectively). The vanilloid agonist sites (red circles) are located in intracellular loops and in the fourth transmembrane domain. It is interesting to note that both heat and proton detection are mediated by specific residues located in extracellular protein loops. Adapted from Refs. 81-83.
Presumptive mechanisms of action of n-3 fatty acids in TRP channels. A, n-3 fatty acids can compete with arachidonic acid and other lipids for PLA2. n-3 fatty acids can subsequently act directly on TRP channels or after their oxygenation by lipoxygenases. B, n-3 fatty acids can increase membrane elasticity and alter the biophysical properties of TRP channels. C, n-3 fatty acids could also exert their effects on Gαq-coupled receptors, which in turn activate phospholipase C (PLC), with subsequent DAG and IP3 release. DAG may activate several TRP channels and IP3 could promote the depletion of intracellular Ca2+ stores, which may also impact the modulation of TRP channels. D, Resolvins may also bind to the Gαi-coupled receptor ChemR23, which can modulate several pathways that modulate TRP channel function. TRP = transient receptor potential; PLA2 = phospholipase 2; DAG = diacylglycerol; IP3 = inositol triphosphate.
1. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000; 1: 11-21.
2. Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, et al. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell 2002; 9: 229-231.
3. Clapham DE. TRP channels as cellular sensors. Nature 2003; 426: 517-524.
4. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol 2006; 68: 619-647.
5. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 2007; 76: 387-417.
6. Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol 2009; 75: 1262-1279.
7. Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium 2005; 38: 233-252.
8. Minke B. TRP channels and Ca2+ signaling. Cell Calcium 2006; 40: 261-275.
9. Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol 2007; 581: 631-647.
10. Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem 2005; 280: 6138-6148.
11. Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci 2009; 12: 1300-1307.
12. Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003; 115: 863-877.
13. Leonelli M, Martins DO, Britto LR. TRPV1 receptors are involved in protein nitration and Muller cell reaction in the acutely axotomized rat retina. Exp Eye Res 2010; 91: 755-768.
14. Balzer M, Lintschinger B, Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res 1999; 42: 543-549.
15. Groschner K, Rosker C, Lukas M. Role of TRP channels in oxidative stress. Novartis Found Symp 2004; 258: 222-230.
16. Medvedeva YV, Kim MS, Usachev YM. Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci 2008; 28: 5295-5311.
17. Lam PM, Hainsworth AH, Smith GD, Owen DE, Davies J, Lambert DG. Activation of recombinant human TRPV1 receptors expressed in SH-SY5Y human neuroblastoma cells increases [Ca(2+)](i), initiates neurotransmitter release and promotes delayed cell death. J Neurochem 2007; 102: 801-811.
18. Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, et al. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem 2007; 102: 977-990.
19. Nilius B, Owsianik G. Transient receptor potential channelopathies. Pflugers Arch 2010; 460: 437-450.
20. Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochim Biophys Acta 2007; 1772: 851-858.
21. Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev 2010; 62: 381-404.
22. Desai BN, Clapham DE. TRP channels and mice deficient in TRP channels. Pflugers Arch 2005; 451: 11-18.
23. Yamamoto S, Wajima T, Hara Y, Nishida M, Mori Y. Transient receptor potential channels in Alzheimer"s disease. Biochim Biophys Acta 2007; 1772: 958-967.
24. Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J Biol Chem 2005; 280: 2132-2140.
25. Hermosura MC, Nayakanti H, Dorovkov MV, Calderon FR, Ryazanov AG, Haymer DS, et al. A TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc Natl Acad Sci U S A 2005; 102: 11510-11515.
26. Nilius B. TRP channels in disease. Biochim Biophys Acta 2007; 1772: 805-812.
27. Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med 2009; 15: 89-100.
28. Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010; 47: 122-129.
29. Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg 1999; 91: 737-743.
30. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001; 411: 590-595.
31. Fleig A, Penner R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci 2004; 25: 633-639.
32. Silverman HS, Di LF, Hui RC, Miyata H, Sollott SJ, Hanford RG, et al. Regulation of intracellular free Mg2+ and contraction in single adult mammalian cardiac myocytes. Am J Physiol 1994; 266: C222-C233.
33. Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 2007; 27: 1129-1138.
34. Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med 2005; 38: 1433-1444.
35. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 2002; 9: 163-173.
36. Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 2006; 101: 66-76.
37. Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 2004; 286: C129-C137.
38. Kuhn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch 2005; 451: 212-219.
39. Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 2001; 3: 193-197.
40. Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 1990; 87: 1620-1624.
41. Leonelli M, Martins DO, Britto LR. TRPV1 receptors modulate retinal development. Int J Dev Neurosci 2011; 29: 405-413.
42. Leonelli M, Martins DO, Kihara AH, Britto LR. Ontogenetic expression of the vanilloid receptors TRPV1 and TRPV2 in the rat retina. Int J Dev Neurosci 2009; 27: 709-718.
43. Leonelli M, Britto LR, Chaves GP, Torrao AS. Developmental expression of cannabinoid receptors in the chick retinotectal system. Brain Res Dev Brain Res 2005; 156: 176-182.
44. Xing J, Li J. TRPV1 receptor mediates glutamatergic synaptic input to dorsolateral periaqueductal gray (dl-PAG) neurons. J Neurophysiol 2007; 97: 503-511.
45. Matta JA, Miyares RL, Ahern GP. TRPV1 is a novel target for omega-3 polyunsaturated fatty acids. J Physiol 2007; 578: 397-411.
46. Bruno MJ, Koeppe RE, Andersen OS. Docosahexaenoic acid alters bilayer elastic properties. Proc Natl Acad Sci U S A 2007; 104: 9638-9643.
47. Andersen OS, Koeppe RE. Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 2007; 36: 107-130.
48. Parnas M, Peters M, Minke B. Linoleic acid inhibits TRP channels with intrinsic voltage sensitivity: Implications on the mechanism of linoleic acid action. Channels 2009; 3: 164-166.
49. Andersson DA, Nash M, Bevan S. Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J Neurosci 2007; 27: 3347-3355.
50. Lee TH, Mencia-Huerta JM, Shih C, Corey EJ, Lewis RA, Austen KF. Effects of exogenous arachidonic, eicosapentaenoic, and docosahexaenoic acids on the generation of 5-lipoxygenase pathway products by ionophore-activated human neutrophils. J Clin Invest 1984; 74: 1922-1933.
51. Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003; 424: 434-438.
52. Hu HZ, Xiao R, Wang C, Gao N, Colton CK, Wood JD, et al. Potentiation of TRPV3 channel function by unsaturated fatty acids. J Cell Physiol 2006; 208: 201-212.
53. Materazzi S, Nassini R, Andre E, Campi B, Amadesi S, Trevisani M, et al. Cox-dependent fatty acid metabolites cause pain through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 2008; 105: 12045-12050.
54. Sun FF, McGuire JC, Metzler CM. The effect of substrate availability on the metabolism of arachidonic acid in human platelets. Prog Lipid Res 1981; 20: 275-278.
55. Madani S, Hichami A, Legrand A, Belleville J, Khan NA. Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. FASEB J 2001; 15: 2595-2601.
56. McGahon BM, Martin DS, Horrobin DF, Lynch MA. Age-related changes in synaptic function: analysis of the effect of dietary supplementation with omega-3 fatty acids. Neuroscience 1999; 94: 305-314.
57. Kotani S, Sakaguchi E, Warashina S, Matsukawa N, Ishikura Y, Kiso Y, et al. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci Res 2006; 56: 159-164.
58. Okaichi Y, Ishikura Y, Akimoto K, Kawashima H, Toyoda-Ono Y, Kiso Y, et al. Arachidonic acid improves aged rats" spatial cognition. Physiol Behav 2005; 84: 617-623.
59. Salem N Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 2001; 36: 945-959.
60. Tsukada H, Kakiuchi T, Fukumoto D, Nishiyama S, Koga K. Docosahexaenoic acid (DHA) improves the age-related impairment of the coupling mechanism between neuronal activation and functional cerebral blood flow response: a PET study in conscious monkeys. Brain Res 2000; 862: 180-186.
61. Kim HY, Akbar M, Lau A, Edsall L. Inhibition of neuronal apoptosis by docosahexaenoic acid (22:6n-3). Role of phosphatidylserine in antiapoptotic effect. J Biol Chem 2000; 275: 35215-35223.
62. Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem 2003; 278: 11303-11311.
63. Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003; 422: 173-176.
64. Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem 2003; 278: 29031-29040.
65. van Rossum DB, Patterson RL. PKC and PLA2: probing the complexities of the calcium network. Cell Calcium 2009; 45: 535-545.
66. Montell C. The TRP superfamily of cation channels. Sci STKE 2005; 2005: re3.
67. Ma D, Tao B, Warashina S, Kotani S, Lu L, Kaplamadzhiev DB, et al. Expression of free fatty acid receptor GPR40 in the central nervous system of adult monkeys. Neurosci Res 2007; 58: 394-401.
68. Ma D, Lu L, Boneva NB, Warashina S, Kaplamadzhiev DB, Mori Y, et al. Expression of free fatty acid receptor GPR40 in the neurogenic niche of adult monkey hippocampus. Hippocampus 2008; 18: 326-333.
69. Moore SA, Yoder E, Murphy S, Dutton GR, Spector AA. Astrocytes, not neurons, produce docosahexaenoic acid (22:6 omega-3) and arachidonic acid (20:4 omega-6). J Neurochem 1991; 56: 518-524.
70. Boneva NB, Kaplamadzhiev DB, Sahara S, Kikuchi H, Pyko IV, Kikuchi M, et al. Expression of fatty acid-binding proteins in adult hippocampal neurogenic niche of postischemic monkeys. Hippocampus 2011; 21: 162-171.
71. Ma D, Zhang M, Larsen CP, Xu F, Hua W, Yamashima T, et al. DHA promotes the neuronal differentiation of rat neural stem cells transfected with GPR40 gene. Brain Res 2010; 1330: 1-8.
72. Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol 2007; 28: 176-183.
73. Bang S, Yoo S, Yang TJ, Cho H, Kim YG, Hwang SW. Resolvin D1 attenuates activation of sensory transient receptor potential channels leading to multiple anti-nociception. Br J Pharmacol 2010; 161: 707-720.
74. Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med 2010; 16: 592-7, 1p.
75. Tai Y, Feng S, Du W, Wang Y. Functional roles of TRPC channels in the developing brain. Pflugers Arch 2009; 458: 283-289.
76. Martorana A, Giampa C, DeMarch Z, Viscomi MT, Patassini S, Sancesario G, et al. Distribution of TRPC1 receptors in dendrites of rat substantia nigra: a confocal and electron microscopy study. Eur J Neurosci 2006; 24: 732-738.
77. Colquhoun A. Lipids, mitochondria and cell death: implications in neuro-oncology. Mol Neurobiol 2010; 42: 76-88.
78. Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci 2008; 1147: 53-60.
79. Engstrom K, Saldeen AS, Yang B, Mehta JL, Saldeen T. Effect of fish oils containing different amounts of EPA, DHA, and antioxidants on plasma and brain fatty acids and brain nitric oxide synthase activity in rats. Ups J Med Sci 2009; 114: 206-213.
80. Tegeder I, Scheving R, Wittig I, Geisslinger G. SNO-ing at the nociceptive synapse? Pharmacol Rev 2011; 63: 366-389.
81. Kauer JA, Gibson HE. Hot flash: TRPV channels in the brain. Trends Neurosci 2009; 32: 215-224.
82. Rohacs T, Thyagarajan B, Lukacs V. Phospholipase C mediated modulation of TRPV1 channels. Mol Neurobiol 2008; 37: 153-163.
83. Jordt SE, Julius D. Molecular basis for species-specific sensitivity to "hot" chili peppers. Cell 2002; 108: 421-430.
The laboratory of L.R.G. Britto is supported by grants from FAPESP and CNPq. M. Leonelli and M.F.R. Graciano are supported by postdoctoral fellowships from FAPESP.
Address for correspondence: M. Leonelli, Departamento de Fisiologia e Biofísica, Instituto de Ciências Biomédicas, USP, Av. Prof. Lineu Prestes, 1524, Sala 239, 05508-000 São Paulo, SP, Brasil. Fax: +55-11-3091-7426. E-mail: mauroleonelli@yahoo.com.br
Received April 27, 2011. Accepted September 8, 2011. Available online September 23, 2011. Published November 14, 2011.
The Brazilian Journal of Medical and Biological Research is partially financed by






All the contents of this journal, except where otherwise noted, is licensed under a Creative Commons Attribution License
- 1. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000; 1: 11-21.
- 2. Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, et al. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell 2002; 9: 229-231.
- 3. Clapham DE. TRP channels as cellular sensors. Nature 2003; 426: 517-524.
- 4. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol 2006; 68: 619-647.
- 5. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 2007; 76: 387-417.
- 6. Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol 2009; 75: 1262-1279.
- 7. Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium 2005; 38: 233-252.
- 8. Minke B. TRP channels and Ca2+ signaling. Cell Calcium 2006; 40: 261-275.
- 9. Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol 2007; 581: 631-647.
- 10. Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem 2005; 280: 6138-6148.
- 11. Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci 2009; 12: 1300-1307.
- 12. Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003; 115: 863-877.
- 13. Leonelli M, Martins DO, Britto LR. TRPV1 receptors are involved in protein nitration and Muller cell reaction in the acutely axotomized rat retina. Exp Eye Res 2010; 91: 755-768.
- 14. Balzer M, Lintschinger B, Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res 1999; 42: 543-549.
- 15. Groschner K, Rosker C, Lukas M. Role of TRP channels in oxidative stress. Novartis Found Symp 2004; 258: 222-230.
- 16. Medvedeva YV, Kim MS, Usachev YM. Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci 2008; 28: 5295-5311.
- 17. Lam PM, Hainsworth AH, Smith GD, Owen DE, Davies J, Lambert DG. Activation of recombinant human TRPV1 receptors expressed in SH-SY5Y human neuroblastoma cells increases [Ca(2+)](i), initiates neurotransmitter release and promotes delayed cell death. J Neurochem 2007; 102: 801-811.
- 18. Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, et al. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem 2007; 102: 977-990.
- 19. Nilius B, Owsianik G. Transient receptor potential channelopathies. Pflugers Arch 2010; 460: 437-450.
- 20. Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochim Biophys Acta 2007; 1772: 851-858.
- 21. Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev 2010; 62: 381-404.
- 22. Desai BN, Clapham DE. TRP channels and mice deficient in TRP channels. Pflugers Arch 2005; 451: 11-18.
- 23. Yamamoto S, Wajima T, Hara Y, Nishida M, Mori Y. Transient receptor potential channels in Alzheimer"s disease. Biochim Biophys Acta 2007; 1772: 958-967.
- 24. Bollimuntha S, Singh BB, Shavali S, Sharma SK, Ebadi M. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J Biol Chem 2005; 280: 2132-2140.
- 25. Hermosura MC, Nayakanti H, Dorovkov MV, Calderon FR, Ryazanov AG, Haymer DS, et al. A TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc Natl Acad Sci U S A 2005; 102: 11510-11515.
- 26. Nilius B. TRP channels in disease. Biochim Biophys Acta 2007; 1772: 805-812.
- 27. Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med 2009; 15: 89-100.
- 28. Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010; 47: 122-129.
- 29. Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg 1999; 91: 737-743.
- 30. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001; 411: 590-595.
- 31. Fleig A, Penner R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci 2004; 25: 633-639.
- 32. Silverman HS, Di LF, Hui RC, Miyata H, Sollott SJ, Hanford RG, et al. Regulation of intracellular free Mg2+ and contraction in single adult mammalian cardiac myocytes. Am J Physiol 1994; 266: C222-C233.
- 33. Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 2007; 27: 1129-1138.
- 34. Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med 2005; 38: 1433-1444.
- 35. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 2002; 9: 163-173.
- 36. Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 2006; 101: 66-76.
- 37. Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 2004; 286: C129-C137.
- 38. Kuhn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch 2005; 451: 212-219.
- 39. Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 2001; 3: 193-197.
- 40. Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 1990; 87: 1620-1624.
- 41. Leonelli M, Martins DO, Britto LR. TRPV1 receptors modulate retinal development. Int J Dev Neurosci 2011; 29: 405-413.
- 42. Leonelli M, Martins DO, Kihara AH, Britto LR. Ontogenetic expression of the vanilloid receptors TRPV1 and TRPV2 in the rat retina. Int J Dev Neurosci 2009; 27: 709-718.
- 43. Leonelli M, Britto LR, Chaves GP, Torrao AS. Developmental expression of cannabinoid receptors in the chick retinotectal system. Brain Res Dev Brain Res 2005; 156: 176-182.
- 44. Xing J, Li J. TRPV1 receptor mediates glutamatergic synaptic input to dorsolateral periaqueductal gray (dl-PAG) neurons. J Neurophysiol 2007; 97: 503-511.
- 45. Matta JA, Miyares RL, Ahern GP. TRPV1 is a novel target for omega-3 polyunsaturated fatty acids. J Physiol 2007; 578: 397-411.
- 46. Bruno MJ, Koeppe RE, Andersen OS. Docosahexaenoic acid alters bilayer elastic properties. Proc Natl Acad Sci U S A 2007; 104: 9638-9643.
- 47. Andersen OS, Koeppe RE. Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 2007; 36: 107-130.
- 48. Parnas M, Peters M, Minke B. Linoleic acid inhibits TRP channels with intrinsic voltage sensitivity: Implications on the mechanism of linoleic acid action. Channels 2009; 3: 164-166.
- 49. Andersson DA, Nash M, Bevan S. Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J Neurosci 2007; 27: 3347-3355.
- 50. Lee TH, Mencia-Huerta JM, Shih C, Corey EJ, Lewis RA, Austen KF. Effects of exogenous arachidonic, eicosapentaenoic, and docosahexaenoic acids on the generation of 5-lipoxygenase pathway products by ionophore-activated human neutrophils. J Clin Invest 1984; 74: 1922-1933.
- 51. Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003; 424: 434-438.
- 52. Hu HZ, Xiao R, Wang C, Gao N, Colton CK, Wood JD, et al. Potentiation of TRPV3 channel function by unsaturated fatty acids. J Cell Physiol 2006; 208: 201-212.
- 53. Materazzi S, Nassini R, Andre E, Campi B, Amadesi S, Trevisani M, et al. Cox-dependent fatty acid metabolites cause pain through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 2008; 105: 12045-12050.
- 54. Sun FF, McGuire JC, Metzler CM. The effect of substrate availability on the metabolism of arachidonic acid in human platelets. Prog Lipid Res 1981; 20: 275-278.
- 55. Madani S, Hichami A, Legrand A, Belleville J, Khan NA. Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. FASEB J 2001; 15: 2595-2601.
- 56. McGahon BM, Martin DS, Horrobin DF, Lynch MA. Age-related changes in synaptic function: analysis of the effect of dietary supplementation with omega-3 fatty acids. Neuroscience 1999; 94: 305-314.
- 57. Kotani S, Sakaguchi E, Warashina S, Matsukawa N, Ishikura Y, Kiso Y, et al. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci Res 2006; 56: 159-164.
- 58. Okaichi Y, Ishikura Y, Akimoto K, Kawashima H, Toyoda-Ono Y, Kiso Y, et al. Arachidonic acid improves aged rats" spatial cognition. Physiol Behav 2005; 84: 617-623.
- 59. Salem N Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 2001; 36: 945-959.
- 60. Tsukada H, Kakiuchi T, Fukumoto D, Nishiyama S, Koga K. Docosahexaenoic acid (DHA) improves the age-related impairment of the coupling mechanism between neuronal activation and functional cerebral blood flow response: a PET study in conscious monkeys. Brain Res 2000; 862: 180-186.
- 61. Kim HY, Akbar M, Lau A, Edsall L. Inhibition of neuronal apoptosis by docosahexaenoic acid (22:6n-3). Role of phosphatidylserine in antiapoptotic effect. J Biol Chem 2000; 275: 35215-35223.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
01 Nov 2011 -
Date of issue
Nov 2011