Abstract
Progressive myelopathies can be secondary to inborn errors of metabolism (IEM) such as mucopolysaccharidosis, mucolipidosis, and adrenomyeloneuropathy. The available scale, Japanese Orthopaedic Association (JOA) score, was validated only for degenerative vertebral diseases. Our objective is to propose and validate a new scale addressing progressive myelopathies and to present validating data for JOA in these diseases. A new scale, Severity Score System for Progressive Myelopathy (SSPROM), covering motor disability, sphincter dysfunction, spasticity, and sensory losses. Inter- and intra-rater reliabilities were measured. External validation was tested by applying JOA, the Expanded Disability Status Scale (EDSS), the Barthel index, and the Osame Motor Disability Score. Thirty-eight patients, 17 with adrenomyeloneuropathy, 3 with mucopolysaccharidosis I, 3 with mucopolysaccharidosis IV, 2 with mucopolysaccharidosis VI, 2 with mucolipidosis, and 11 with human T-cell lymphotropic virus type-1 (HTLV-1)-associated myelopathy participated in the study. The mean ± SD SSPROM and JOA scores were 74.6 ± 11.4 and 12.4 ± 2.3, respectively. Construct validity for SSPROM (JOA: r = 0.84, P < 0.0001; EDSS: r = -0.83, P < 0.0001; Barthel: r = 0.56, P < 0.002; Osame: r = -0.94, P < 0.0001) and reliability (intra-rater: r = 0.83, P < 0.0001; inter-rater: r = 0.94, P < 0.0001) were demonstrated. The metric properties of JOA were similar to those found in SSPROM. Several clinimetric requirements were met for both SSPROM and JOA scales. Since SSPROM has a wider range, it should be useful for follow-up studies on IEM myelopathies.
Japanese Orthopaedic Association; Severity Score System for Progressive Myelopathy; Mucopolysaccharidosis; Mucolipidosis; Adrenomyeloneuropathy; Progressive myelopathies
Braz J Med Biol Res, July 2012, Volume 45(7) 565-572
Severity score system for progressive myelopathy: development and validation of a new clinical scale
R.M. Castilhos1,4, D. Blank4, C.B.O. Netto4, C.F.M. Souza4, L.N.T. Fernandes3, I.V.D. Schwartz1,2,4, R. Giugliani1,2,4 and  Correspondence and Footnotes
Correspondence and Footnotes
 L.B. Jardim1,3,4
L.B. Jardim1,3,4
1Programa de Pós-Graduação em Ciências Médicas, Porto Alegre, RS, Brasil
2Departamento de Genética e Biologia Molecular, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil
3Departamento de Medicina Interna, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil
4Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Porto Alegre, RS, Brasil
 Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Progressive myelopathies can be secondary to inborn errors of metabolism (IEM) such as mucopolysaccharidosis, mucolipidosis, and adrenomyeloneuropathy. The available scale, Japanese Orthopaedic Association (JOA) score, was validated only for degenerative vertebral diseases. Our objective is to propose and validate a new scale addressing progressive myelopathies and to present validating data for JOA in these diseases. A new scale, Severity Score System for Progressive Myelopathy (SSPROM), was constructed covering motor disability, sphincter dysfunction, spasticity, and sensory losses. Inter- and intra-rater reliabilities were measured. External validation was tested by applying JOA, the Expanded Disability Status Scale (EDSS), the Barthel index, and the Osame Motor Disability Score. Thirty-eight patients, 17 with adrenomyeloneuropathy, 3 with mucopolysaccharidosis I, 3 with mucopolysaccharidosis IV, 2 with mucopolysaccharidosis VI, 2 with mucolipidosis, and 11 with human T-cell lymphotropic virus type-1 (HTLV-1)-associated myelopathy participated in the study. The mean ± SD SSPROM and JOA scores were 74.6 ± 11.4 and 12.4 ± 2.3, respectively. Construct validity for SSPROM (JOA: r = 0.84, P < 0.0001; EDSS: r = -0.83, P < 0.0001; Barthel: r = 0.56, P < 0.002; Osame: r = -0.94, P < 0.0001) and reliability (intra-rater: r = 0.83, P < 0.0001; inter-rater: r = 0.94, P < 0.0001) were demonstrated. The metric properties of JOA were similar to those found in SSPROM. Several clinimetric requirements were met for both SSPROM and JOA scales. Since SSPROM has a wider range, it should be useful for follow-up studies on IEM myelopathies.
Key words: Japanese Orthopaedic Association; Severity Score System for Progressive Myelopathy; Mucopolysaccharidosis; Mucolipidosis; Adrenomyeloneuropathy; Progressive myelopathies
Introduction
Lesions of the spinal cord are characterized by sensory, motor and autonomic changes. Clinical presentations vary, including any combination of strength and sensory loss, hyperactive stretch reflexes and sphincter abnormalities. The resulting symptoms will depend not only upon the location of the damage, but also upon the rapidity of the progression of the underlying disease. As a result, myelopathies can be divided into static and progressive disorders, with very different clinical courses and etiologies (1).
Among other causes, some inborn errors of metabolism (IEM) may cause progressive myelopathies, such as mucopolysaccharidosis (MPS) (2,3), mucolipidoses (ML) (4), and adrenomyeloneuropathy (AMN) (5). At least two mechanisms might be involved.
In MPS and ML, spinal cord compression is the main underlying mechanism, which can be secondary to a) vertebral abnormalities (hypoplasia or absence of the odontoid process, or subluxation due to ligamentous laxity), or b) infiltration of surrounding soft tissue structures by undegraded glycosaminoglycans, also termed pachymeningitis cervicalis (6,7). In contrast, the myelopathy associated with AMN is primarily due to axonal degeneration, with spinal cord atrophy (8).
Several scales have been developed to measure the stage of disability or disease in a number of pathologies involving the spinal cord. Some scales have addressed non-progressive diseases, such as vascular or traumatic lesions. This is the case for the American Spinal Injury Association (ASIA) scale (9). Other scales were designed to encompass some aspects of progressive spinal cord diseases, such as those related to autoimmune, viral or compressive etiologies (10,11). Nevertheless, none of them were specifically developed for the progressive involvement of both the ascending and descending tracts of the spinal cord in IEM. The well-known Japanese Orthopaedic Association (JOA) score (12) was developed to evaluate patients with degenerative cervical compression. JOA is presently being used in clinical trials on MPS (Dickson P, personal communication), although the disability profile of these patients is most probably different from that of patients with degenerative vertebral diseases. The widely used Kurtzke Expanded Disability Status Scale (EDSS) and Kurtzke Functional Systems Scores (FSS) were developed to follow patients with multiple sclerosis. For this reason, EDSS and FSS include aspects not related to the spinal cord, such as the assessment of cerebellar, brain stem, visual, and cerebral functions. Although they have never been validated for these purposes, EDSS and FSS have been used to follow patients with human T-cell lymphotropic virus type-1 (HTLV-1) myelopathy (13,14). We have recently used the EDSS to follow the progress of MPS I and MPS VI patients receiving intrathecal therapy (15,16).
In order to deal with this deficiency, we propose a new clinical scale tailored to progressive myelopathies in general, aimed at providing better responses, and present validation data for the JOA in these disorders. We report the reliability and validity of the new scale in patients with myelopathies caused by MPS, AMN and HTLV-1 infection.
Material and Methods
Construction of the new scale
A disease severity scoring system was constructed, called the Severity Score System for Progressive Myelopathy (SSPROM) (Supporting Information). We aimed to include most of the symptoms of spinal cord long tract involvement such as weakness, sensory loss, hyperreflexia, spasticity, and sphincter dysfunction. At the same time, in order to facilitate acceptance and feasibility, several partial scales that are well established in clinical practice were incorporated into the SSPROM. For these two reasons, SSPROM may be seen as a "scoring system". The SSPROM ranged from zero (total disability) to 100 (normal) and covered the following domains: "motor disability reported" and "examined", "sphincter dysfunction", and "presence of spasticity and hyperreflexia", and of "sensory losses".
Two domains are subjective and rely on patient (or relative) information: the motor disability and the sphincter dysfunction reported. The "motor disability reported" domain was based on the queries presented by the Overall Disability Sum Score (ODSS) (17). The "sphincter dysfunction" domain was adapted from the related subscore of FSS (11).
The other domains are related to quantitative assessment of the standardized neurological examination. The "motor examined domain" was adapted from the widely used Medical Research Council scale (18). The "presence of spasticity and hyperreflexia" domain was based on the Ashworth scale (19), to which we added atony and two- or four-limb hyperreflexia as other possible findings. All scales we used to construct the new scale are very well known and contain items suitable for measuring myelopathy. All were adapted to be in a descending order. In contrast, the "sensory loss" domain was originally developed to be used in the SSPROM.
All items were weighted, so that the contributions of each system would be as follows: motor strength, 50%; tonus and stretch reflexes, 10%; sphincter function, 20%, and sensory system, 20%. All of these domains are summed to obtain a final score (0 to 100) (SSPROM) (Supporting Information).
Patients and validation procedures
The study was performed between January 2009 and January 2010, and was approved by the Ethics Committee of our institution (Hospital de Clínicas de Porto Alegre). Informed consent was obtained from all participants, or from their parents or guardians. SSPROM was initially addressed to myelopathies due to certain IEM such as MPS and AMN. Since these patients are quite rare, patients with HTLV-1 infection were also included in the study, due to the progressive nature of this disease. Exclusion criteria were the presence of cognitive involvement or a central nervous system disease causing neurologic dysfunction other than myelopathy. Patients were invited for two consecutive visits to our outpatient clinics. During the first visit, two examiners (RMC and DB) evaluated each patient in order to test inter-rater correlations. Ratings for the new scale and for JOA were generated independently by both investigators, and each examiner was blind to the other’s score. An additional evaluation (on the occasion of the second visit) was performed by one of the examiners (RMC) 30 to 45 days after the first examination in order to evaluate intra-rater reliability of SSPROM and JOA. To explore criterion validity (20,21), on the occasion of the second visit we applied the following scales: EDSS, the Barthel index (22), Osame Motor Disability Score (23), and JOA.
Statistical analysis
To test correlations among measures, Pearson’s coefficient was employed for continuous variables, and the Spearman rank test was used for ordinal or not normally distributed variables. Inter- and intra-rater reliabilities were determined by intraclass correlation (24,25). All tests were two-tailed; P values less than 0.05 were considered to be statistically significant. Analyses were performed using SPSS 18.0 for Windows.
Results
Sample characteristics
Thirty-eight (18 males) patients with progressive myelopathies had accepted our invitation to participate in the study by January 2010. There were 17 cases of AMN, 3 of MPS I, 3 of MPS IV, 2 of MPS VI, 2 of ML, and 11 of HTLV-1 infection. Patient age, disease onset and disease duration are shown in Table 1.
Analysis of SSPROM
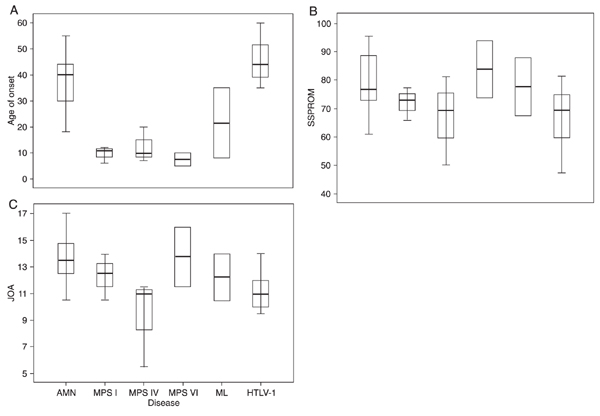
SSPROM was easy to administer and required an average of 15 min per patient. The mean (range) SSPROM score for the present sample was 74.61 (47.5 to 95.5). Scaling characteristics are summarized in Table 2. The present sample of patients showed a ceiling effect in SSPROM as well as in the JOA and Barthel scales, reflecting the inclusion of mild to moderate cases (Figure 1).
Distribution of scores among the various diseases under study. There were significant differences in mean ± SD SSPROM scores according to the disease, with worse scores for HTLV-1 and MPS IV (67 ± 15.8 and 67 ± 11, respectively, P = 0.038, ANOVA; Figure 2). All the scales under study were evaluated separately for each disease, namely: AMN, MPS/ML, and HTLV-1. In the MPS/ML subgroup, an association between Barthel scale and age was found (r = 0.89, P = 0.04).
Reliability: inter- and intra-rater tests. The intra-class correlation between the two examiners (blind to each other) was very high (r = 0.96, CI = 93-98). Moreover, a high intra-class correlation was also found between the first and the second SSPROM, performed by the same examiner, with a 30- to 45-day interval (r = 0.84, CI = 0.64-0.90; Table 2).
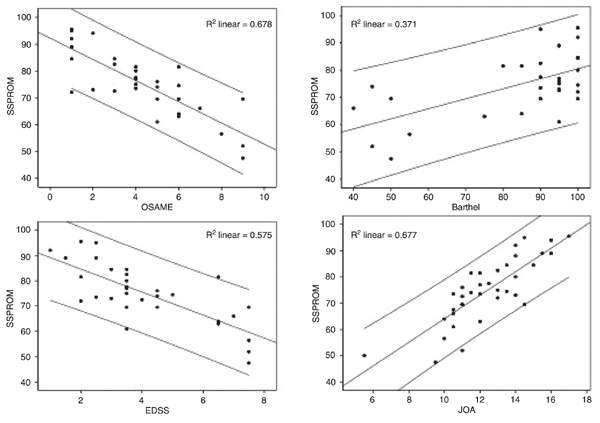
External validity. By using the Spearman rank test, SSPROM showed a good correlation with JOA (r = 0.81, P < 0.0001), EDSS (r = -0.85, P < 0.0001) and the Osame Motor Disability Score (r = -0.89, P < 0.0001). Although significant (P < 0.001), the correlation with the Barthel scale was only moderate (r = 0.58; Table 2 and Figure 3).
Since six different disorders were included in this study, the main accepted factors of disease severity, such as disease duration and stage reached, could not be used to test external validity. Although SSPROM seemed to be associated with disease duration for the two largest groups of patients - AMN and HTLV-1 -, there was no statistical significance (Figure 4).
Analysis of JOA
JOA was also easy to administer and required an average of 10 min per patient. The mean (range) JOA score in the present sample was 12.4 (6 to 17). Scaling characteristics are illustrated in Table 2 and Figure 1, and were quite similar to the SSPROM characteristics.
MPS IV patients presented more severe mean ± SD JOA scores than the remaining ones (9.3 ± 3, P = 0.035, ANOVA); JOA scores for HTLV-1 patients did not differ from those of the MPS and ML subgroups, as observed for the SSPROM scores (Figure 3).
JOA inter- and intra-rater tests showed high reliability, similar to those found for SSPROM. External validation of JOA included good correlations with the Barthel, EDSS and Osame scales (Table 2). As already stated, the main accepted factors of disease severity, such as disease duration and stage reached, could not be used to test external validity in the overall sample due to the heterogeneity of the diseases under study. Therefore, these parameters were compared to JOA inside the disease subgroups HTLV-1, MPS/ML, and AMN. In the AMN subgroup, JOA was associated with disease duration (r = -0.59, P = 0.02) (Figure 4).
Distribution of the scores obtained in this study with the (I) SSPROM, (II) JOA, (III) Barthel, (IV) EDSS, and (V) Osame scales. Lower row: score distribution according to each disease under study. A = adrenomyeloneuropathy (AMN); B = mucopolysaccharidosis type I (MPS I), C = MPS type IV (MPS IV); D = MPS type VI (MPS VI); E = mucolipidosis (ML); F = human T-cell lymphotropic virus type-1-associated myelopathy (HTLV-1). SSPROM = Severity Score System for Progressive Myelopathy; JOA = Japanese Orthopaedic Association score; EDSS = Kurtzke Expanded Disability Status Scale; OSAME = Osame Motor Disability Score.
Distribution of (A) age at onset, (B) SSPROM and (C) JOA scores according to the different diseases under study. Data are reported as the mean, SD, and range. SSPROM = Severity Score System for Progressive Myelopathy; JOA = Japanese Orthopaedic Association score; AMN = adrenomyeloneuropathy; MPS = mucopolysaccharidosis types I, IV and VI; ML = mucolipidosis; HTLV-1 = human T-cell lymphotropic virus type-1.
Correlations between SSPROM and other scales under study. SSPROM = Severity Score System for Progressive Myelopathy; JOA = Japanese Orthopaedic Association score; EDSS = Kurtzke Expanded Disability Status Scale; OSAME = Osame Motor Disability Score.
Correlations between disease duration and SSPROM in (A) HTLV-1 infection and (B) adrenomyeloneuropathy, and between (C) disease duration and JOA for adrenomyeloneuropathy. SSPROM = Severity Score System for Progressive Myelopathy; JOA = Japanese Orthopaedic Association score; AMN = adrenomyeloneuropathy; HTLV-1 = human T cell lymphotropic virus type-1.
Discussion
Myelopathy is an important cause of disability in some metabolic diseases. As far as we know, no scale has been designed to measure the disability and disease progression of myelopathy in these disorders. Therefore, we planned to construct a severity scale - SSPROM - with a wide range, also including disability information, addressing an important population that is not properly represented by other scales.
The JOA scale was reported in 2001, and was developed to measure the myelopathy caused by cervical spondylotic myelopathy, a disease that usually affects individuals aged 50 years or older (12). However, JOA was not validated for AMN or for glycosaminoglycan storage diseases (26), and has a restricted range, between 2 and 17 points, which may raise some concerns about its responsiveness during a hypothetical treatment. After all, new therapies for the metabolic causes of myelopathies are being developed (15,16,27) and a reliable and validated method to measure efficacy is needed.
The present study investigated some fundamental metric properties of SSPROM, and our results suggest that this proposed scale was feasible and consistent. The items used in the scale follow the procedural steps of a standard neurological examination, which increases its content validity and acceptability. By adapting SSPROM to already well-accepted and widely used measurements, we aimed to construct a reproducible instrument.
Inter- and intra-observer correlations of SSPROM scores were very good, showing within- and between-physician agreement when assessing the same patient on the same date (or after 30-45 days) using SSPROM. SSPROM does not require expensive equipment and is widely available to the medical community in general, and can be applied in an acceptable time. External validation was achieved, since SSPROM correlated very strongly with at least three of the four external variables under study - JOA, Osame, and EDSS. A moderate correlation was found between SSPROM and Barthel, with r = 0.54. Barthel is well known for its strong ceiling effect and low responsiveness in patients with stroke, multiple sclerosis and from neurorehabilitation units (28,29). This is probably because most of its items have only two or three response options (30). Nevertheless, Barthel was never tested in patients with MPS/ML or AMN; the only study that used it in patients with HTLV infection did not measure any clinimetric parameters (31).
One concern that may arise is about the small size of the sample investigated here. Since we were dealing with very rare diseases, recruiting more patients was not an easy task, especially because we needed to exclude patients with cognitive impairment. However, the strong correlations found do reflect external validity and reinforce the utility of SSPROM.
The distribution of scores was skewed towards moderate to mild results in several scales studied. This phenomenon was observed in the SSPROM, JOA and Barthel scores, and probably reflects a selection bias produced by the exclusion of more severely affected patients. Cognitive involvement may be associated with more severe clinical pictures, at least in patients with MPS and ML (3,32). Therefore, JOA and SSPROM can be criticized for not being useful for severe, cognitively affected patients. Both JOA and SSPROM rely on subjective information during sensory examination. This can be circumvented by the application of a "reduced-SSPROM scale" (without the sensory subscore) to additional MPS patients with cognitive involvement. In any case, this, once gain, cannot explain the ceiling effect that occurs in AMN or HTLV-1 patients.
In the present patient sample, the metrical characteristics of JOA were good, and indeed very similar to those found in SSPROM. These results suggest that JOA may be applied to IEM patients, and either procedure (JOA or SSPROM) can be chosen to follow these patients. Since JOA is well established, one may argue about the necessity of introducing a new scale. We can reply that SSPROM was devised to have a high responsiveness to changes in the clinical picture. This characteristic needs to be tested in the future after a good period of time has elapsed to allow changes in the clinical picture to emerge. Since SSPROM differs from JOA in its wide range and because it includes a disability domain, we believe it will be useful for follow-up studies on progressive myelopathies.
References
1. Byrne TN, Waxman SG. Paraplegia and spinal cord syndromes. In: Bradley WG, Daroff RB (Editors), Neurology in clinical practice. Philadelphia: Butterworth Heinemann; 2004. p 351-366.
2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr 2004; 144: S27-S34.
3. Kachur E, Del Maestro R. Mucopolysaccharidoses and spinal cord compression: case report and review of the literature with implications of bone marrow transplantation. Neurosurgery 2000; 47: 223-228.
4. Carl A, Waldman J, Malone A, Blair B. Atlantoaxial instability and myelopathy in mucolipidosis. Spine 1991; 16: 215-217.
5. Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 1997; 120 (Part 8): 1485-1508.
6. Al Sawaf S, Mayatepek E, Hoffmann B. Neurological findings in Hunter disease: pathology and possible therapeutic effects reviewed. J Inherit Metab Dis 2008; 31: 473-480.
7. Parsons VJ, Hughes DG, Wraith JE. Magnetic resonance imaging of the brain, neck and cervical spine in mild Hunter’s syndrome (mucopolysaccharidoses type II). Clin Radiol 1996; 51: 719-723.
8. van Geel BM, Assies J, Wanders RJ, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. J Neurol Neurosurg Psychiatry 1997; 63: 4-14.
9. Maynard FM Jr, Bracken MB, Creasey G, Ditunno JF Jr, Donovan WH, Ducker TB, et al. International Standards for Neurological and Functional Classification of Spinal Cord Injury. American Spinal Injury Association. Spinal Cord 1997; 35: 266-274.
10. Izumo S, Goto I, Itoyama Y, Okajima T, Watanabe S, Kuroda Y, et al. Interferon-alpha is effective in HTLV-I-associated myelopathy: a multicenter, randomized, double-blind, controlled trial. Neurology 1996; 46: 1016-1021.
11. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444-1452.
12. Yonenobu K, Abumi K, Nagata K, Taketomi E, Ueyama K. Interobserver and intraobserver reliability of the Japanese orthopaedic association scoring system for evaluation of cervical compression myelopathy. Spine 2001; 26: 1890-1894.
13. Croda MG, de Oliveira AC, Vergara MP, Bonasser F, Smid J, Duarte AJ, et al. Corticosteroid therapy in TSP/HAM patients: the results from a 10 years open cohort. J Neurol Sci 2008; 269: 133-137.
14. Araujo AQ, Leite AC, Dultra SV, Andrada-Serpa MJ. Progression of neurological disability in HTLV-I-associated myelopathy/tropical spastic paraparesis (HAM/TSP). J Neurol Sci 1995; 129: 147-151.
15. Munoz-Rojas MV, Vieira T, Costa R, Fagondes S, John A, Jardim LB, et al. Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am J Med Genet A 2008; 146A: 2538-2544.
16. Munoz-Rojas MV, Horovitz DD, Jardim LB, Raymundo M, Llerena JC Jr, de Magalhaes TS, et al. Intrathecal administration of recombinant human N-acetylgalactosamine 4-sulfatase to a MPS VI patient with pachymeningitis cervicalis. Mol Genet Metab 2010; 99: 346-350.
17. Merkies IS, Schmitz PI, van der Meche FG, Samijn JP, van Doorn PA. Clinimetric evaluation of a new overall disability scale in immune mediated polyneuropathies. J Neurol Neurosurg Psychiatry 2002; 72: 596-601.
18. Medical Research Council. Aids to the examination of the peripheral nervous system. Memorandum No. 45. London: Her Majesty’s Stationery Office; 1976.
19. Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther 1987; 67: 206-207.
20. Pietrobon R, Coeytaux RR, Carey TS, Richardson WJ, DeVellis RF. Standard scales for measurement of functional outcome for cervical pain or dysfunction: a systematic review. Spine 2002; 27: 515-522.
21. Streiner DL, Norman GR. Health Measurement Scales: a practical guide to their development and use. 4th edn. New York: Oxford University Press; 2008.
22. Mahoney FI, Barthel DW. Functional evaluation: the Barthel Index. Md State Med J 1965; 14: 61-65.
23. Osame M. Review of WHO Kagoshima meeting and diagnostic guidelines for HAM/TSP. In: Blattner WA (Editor), Human retrovirology: HTLV. New York: Raven Press; 1990. p 191-197.
24. Weir JP. Quantifying test-retest reliability using the intraclass correlation coefficient and the SEM. J Strength Cond Res 2005; 19: 231-240.
25. Baumgartner TA. Estimating the stability reliability of a score. Meas Phys Educ Exerc Sci 2000; 4: 175-178.
26. King JT Jr, Tsevat J, Moossy JJ, Roberts MS. Preference-based quality of life measurement in patients with cervical spondylotic myelopathy. Spine 2004; 29: 1271-1280.
27. Kakkis E, McEntee M, Vogler C, Le S, Levy B, Belichenko P, et al. Intrathecal enzyme replacement therapy reduces lysosomal storage in the brain and meninges of the canine model of MPS I. Mol Genet Metab 2004; 83: 163-174.
28. Kwon S, Hartzema AG, Duncan PW, Min-Lai S. Disability measures in stroke: relationship among the Barthel Index, the Functional Independence Measure, and the Modified Rankin Scale. Stroke 2004; 35: 918-923.
29. Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Health Technol Assess 2009; 13: iii, ix-iii, 177.
30. Cano SJ, O’Connor RJ, Thompson AJ, Hobart JC. Exploring disability rating scale responsiveness II: do more response options help? Neurology 2006; 67: 2056-2059.
31. Carod-Artal FJ, Mesquita HM, Ribeiro LS. [Neurological symptoms and disability in HTLV-1 associated myelopathy]. Neurologia 2008; 23: 78-84.
32. Vougioukas VI, Berlis A, Kopp MV, Korinthenberg R, Spreer J, van Velthoven V. Neurosurgical interventions in children with Maroteaux-Lamy syndrome. Case report and review of the literature. Pediatr Neurosurg 2001; 35: 35-38.
Acknowledgments
We are grateful to the patients who participated in this study. We also thank Dr. Patricia I. Dickson, from David Geffen School of Medicine at UCLA, for sharing information about the use of JOA in ongoing clinical trials. Research supported by the Brazilian research agencies FIPE-HCPA, CNPq, CAPES, INAGEMP, and FAPERGS. L.B. Jardim is the recipient of a CNPq fellowship.
Address for correspondence: L.B. Jardim, Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Rua Ramiro Barcelos, 2350, 90035-903 Porto Alegre, RS, Brasil. Fax +55-51-3359-8010. E-mail: ljardim@hcpa.ufrgs.br
Received September 17, 2011. Accepted April 10, 2012. Available online May 11, 2012. Published July 2, 2012.
The Brazilian Journal of Medical and Biological Research is partially financed by






- 1. Byrne TN, Waxman SG. Paraplegia and spinal cord syndromes. In: Bradley WG, Daroff RB (Editors), Neurology in clinical practice Philadelphia: Butterworth Heinemann; 2004. p 351-366.
- 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr 2004; 144: S27-S34.
- 3. Kachur E, Del Maestro R. Mucopolysaccharidoses and spinal cord compression: case report and review of the literature with implications of bone marrow transplantation. Neurosurgery 2000; 47: 223-228.
- 4. Carl A, Waldman J, Malone A, Blair B. Atlantoaxial instability and myelopathy in mucolipidosis. Spine 1991; 16: 215-217.
- 5. Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 1997; 120 (Part 8): 1485-1508.
- 6. Al Sawaf S, Mayatepek E, Hoffmann B. Neurological findings in Hunter disease: pathology and possible therapeutic effects reviewed. J Inherit Metab Dis 2008; 31: 473-480.
- 8. van Geel BM, Assies J, Wanders RJ, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. J Neurol Neurosurg Psychiatry 1997; 63: 4-14.
- 9. Maynard FM Jr, Bracken MB, Creasey G, Ditunno JF Jr, Donovan WH, Ducker TB, et al. International Standards for Neurological and Functional Classification of Spinal Cord Injury. American Spinal Injury Association. Spinal Cord 1997; 35: 266-274.
- 10. Izumo S, Goto I, Itoyama Y, Okajima T, Watanabe S, Kuroda Y, et al. Interferon-alpha is effective in HTLV-I-associated myelopathy: a multicenter, randomized, double-blind, controlled trial. Neurology 1996; 46: 1016-1021.
- 11. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444-1452.
- 12. Yonenobu K, Abumi K, Nagata K, Taketomi E, Ueyama K. Interobserver and intraobserver reliability of the Japanese orthopaedic association scoring system for evaluation of cervical compression myelopathy. Spine 2001; 26: 1890-1894.
- 13. Croda MG, de Oliveira AC, Vergara MP, Bonasser F, Smid J, Duarte AJ, et al. Corticosteroid therapy in TSP/HAM patients: the results from a 10 years open cohort. J Neurol Sci 2008; 269: 133-137.
- 14. Araujo AQ, Leite AC, Dultra SV, Andrada-Serpa MJ. Progression of neurological disability in HTLV-I-associated myelopathy/tropical spastic paraparesis (HAM/TSP). J Neurol Sci 1995; 129: 147-151.
- 15. Munoz-Rojas MV, Vieira T, Costa R, Fagondes S, John A, Jardim LB, et al. Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am J Med Genet A 2008; 146A: 2538-2544.
- 16. Munoz-Rojas MV, Horovitz DD, Jardim LB, Raymundo M, Llerena JC Jr, de Magalhaes TS, et al. Intrathecal administration of recombinant human N-acetylgalactosamine 4-sulfatase to a MPS VI patient with pachymeningitis cervicalis. Mol Genet Metab 2010; 99: 346-350.
- 17. Merkies IS, Schmitz PI, van der Meche FG, Samijn JP, van Doorn PA. Clinimetric evaluation of a new overall disability scale in immune mediated polyneuropathies. J Neurol Neurosurg Psychiatry 2002; 72: 596-601.
- 19. Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther 1987; 67: 206-207.
- 20. Pietrobon R, Coeytaux RR, Carey TS, Richardson WJ, DeVellis RF. Standard scales for measurement of functional outcome for cervical pain or dysfunction: a systematic review. Spine 2002; 27: 515-522.
- 21. Streiner DL, Norman GR. Health Measurement Scales: a practical guide to their development and use 4th edn. New York: Oxford University Press; 2008.
- 22. Mahoney FI, Barthel DW. Functional evaluation: the Barthel Index. Md State Med J 1965; 14: 61-65.
- 23. Osame M. Review of WHO Kagoshima meeting and diagnostic guidelines for HAM/TSP. In: Blattner WA (Editor), Human retrovirology: HTLV New York: Raven Press; 1990. p 191-197.
- 24. Weir JP. Quantifying test-retest reliability using the intraclass correlation coefficient and the SEM. J Strength Cond Res 2005; 19: 231-240.
- 25. Baumgartner TA. Estimating the stability reliability of a score. Meas Phys Educ Exerc Sci 2000; 4: 175-178.
- 26. King JT Jr, Tsevat J, Moossy JJ, Roberts MS. Preference-based quality of life measurement in patients with cervical spondylotic myelopathy. Spine 2004; 29: 1271-1280.
- 27. Kakkis E, McEntee M, Vogler C, Le S, Levy B, Belichenko P, et al. Intrathecal enzyme replacement therapy reduces lysosomal storage in the brain and meninges of the canine model of MPS I. Mol Genet Metab 2004; 83: 163-174.
- 28. Kwon S, Hartzema AG, Duncan PW, Min-Lai S. Disability measures in stroke: relationship among the Barthel Index, the Functional Independence Measure, and the Modified Rankin Scale. Stroke 2004; 35: 918-923.
- 29. Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Health Technol Assess 2009; 13: iii, ix-iii, 177.
- 31. Carod-Artal FJ, Mesquita HM, Ribeiro LS. [Neurological symptoms and disability in HTLV-1 associated myelopathy]. Neurologia 2008; 23: 78-84.
- 32. Vougioukas VI, Berlis A, Kopp MV, Korinthenberg R, Spreer J, van Velthoven V. Neurosurgical interventions in children with Maroteaux-Lamy syndrome. Case report and review of the literature. Pediatr Neurosurg 2001; 35: 35-38.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
22 June 2012 -
Date of issue
July 2012
History
-
Received
17 Sept 2011 -
Accepted
10 Apr 2012