Abstract
Sublethal ischemic preconditioning (IPC) is a powerful inducer of ischemic brain tolerance. However, its underlying mechanisms are still not well understood. In this study, we chose four different IPC paradigms, namely 5 min (5 min duration), 5×5 min (5 min duration, 2 episodes, 15-min interval), 5×5×5 min (5 min duration, 3 episodes, 15-min intervals), and 15 min (15 min duration), and demonstrated that three episodes of 5 min IPC activated autophagy to the greatest extent 24 h after IPC, as evidenced by Beclin expression and LC3-I/II conversion. Autophagic activation was mediated by the tuberous sclerosis type 1 (TSC1)-mTor signal pathway as IPC increased TSC1 but decreased mTor phosphorylation. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and hematoxylin and eosin staining confirmed that IPC protected against cerebral ischemic/reperfusion (I/R) injury. Critically, 3-methyladenine, an inhibitor of autophagy, abolished the neuroprotection of IPC and, by contrast, rapamycin, an autophagy inducer, potentiated it. Cleaved caspase-3 expression, neurological scores, and infarct volume in different groups further confirmed the protection of IPC against I/R injury. Taken together, our data indicate that autophagy activation might underlie the protection of IPC against ischemic injury by inhibiting apoptosis.
Ischemic preconditioning; Autophagy; Apoptosis; Focal cerebral ischemic reperfusion
Introduction
Stroke is well-known for causing death and adult disability (11. Hankey GJ, Jamrozik K, Broadhurst RJ, Forbes S, Burvill PW, Anderson CS, et al. Five-year survival after first-ever stroke and related prognostic factors in the Perth Community Stroke Study. Stroke 2000; 31: 2080-2086, doi: 10.1161/01.STR.31.9.2080.
https://doi.org/10.1161/01.STR.31.9.2080...
). Disclosing the pathological mechanisms and finding potential treatment strategies are major topics attracting concern worldwide. Experiments have shown the potential of several interventions to improve the outcomes of this disease (22. Gosman-Hedstrom G, Claesson L, Klingenstierna U, Carlsson J, Olausson B, Frizell M, et al. Effects of acupuncture treatment on daily life activities and quality of life: a controlled, prospective, and randomized study of acute stroke patients. Stroke 1998; 29: 2100-2108, doi: 10.1161/01.STR.29.10.2100.
https://doi.org/10.1161/01.STR.29.10.210...
3. Naeser MA, Hamblin MR. Potential for transcranial laser or LED therapy to treat stroke, traumatic brain injury, and neurodegenerative disease. Photomed Laser Surg 2011; 29: 443-446, doi: 10.1089/pho.2011.9908.
https://doi.org/10.1089/pho.2011.9908...
-44. Minnerup J, Schabitz WR. Improving outcome after stroke: time to treat new targets. Stroke 2012; 43: 295-296, doi: 10.1161/STROKEAHA.111.642363.
https://doi.org/10.1161/STROKEAHA.111.64...
). Recently, some experimental data applied to clinical study have yielded valuable results. Ischemic preconditioning (IPC) is one of the effective intervening strategies that has attracted extensive attention (55. Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 2012; 79: 1853-1861, doi: 10.1212/WNL.0b013e318271f76a.
https://doi.org/10.1212/WNL.0b013e318271...
6. Pignataro G, Scorziello A, Di RG, Annunziato L. Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J 2009; 276: 46-57, doi: 10.1111/j.1742-4658.2008.06769.x.
https://doi.org/10.1111/j.1742-4658.2008...
7. Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, et al. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke 2004; 35: 2687-2689, doi: 10.1161/01.STR.0000143216.85349.9e.
https://doi.org/10.1161/01.STR.000014321...
8. Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, et al. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke 2004; 35: 616-621, doi: 10.1161/01.STR.0000115767.17923.6A.
https://doi.org/10.1161/01.STR.000011576...
-99. Schaller B, Graf R. Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol 2002; 249: 1503-1511, doi: 10.1007/s00415-002-0933-8.
https://doi.org/10.1007/s00415-002-0933-...
).
The term "ischemic preconditioning" was first introduced by Janoff et al. (1010. Janoff A, Weissmann G, Zweiofach BW, Thomas L. Pathogenesis of experimental shock. IV. Studies on lysosomes in normal and tolerant animals subjected to lethal trauma and endotoxemia. J Exp Med 1962; 116: 451-466, doi: 10.1084/jem.116.4.451.
https://doi.org/10.1084/jem.116.4.451...
) in a study of the shock model, and it is now proposed as a powerful inducer of ischemic tolerance (1111. Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29: 1937-1950, doi: 10.1161/01.STR.29.9.1937.
https://doi.org/10.1161/01.STR.29.9.1937...
). A large amount of evidence points to IPC playing a protective role in ischemic/reperfusion (I/R) models in different organs, including the kidney (1212. Zimmerman RF, Ezeanuna PU, Kane JC, Cleland CD, Kempananjappa TJ, Lucas FL, et al. Ischemic preconditioning at a remote site prevents acute kidney injury in patients following cardiac surgery. Kidney Int 2011; 80: 861-867, doi: 10.1038/ki.2011.156.
https://doi.org/10.1038/ki.2011.156...
), heart (1313. Quarrie R, Cramer BM, Lee DS, Steinbaugh GE, Erdahl W, Pfeiffer DR, et al. Ischemic preconditioning decreases mitochondrial proton leak and reactive oxygen species production in the postischemic heart. J Surg Res 2011; 165: 5-14, doi: 10.1016/j.jss.2010.09.018.
https://doi.org/10.1016/j.jss.2010.09.01...
), and brain (99. Schaller B, Graf R. Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol 2002; 249: 1503-1511, doi: 10.1007/s00415-002-0933-8.
https://doi.org/10.1007/s00415-002-0933-...
, 1111. Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29: 1937-1950, doi: 10.1161/01.STR.29.9.1937.
https://doi.org/10.1161/01.STR.29.9.1937...
). Although protection from stroke morbidity by sublethal ischemic stress is thought to be largely due to tolerance or adjustment, an effective or convincing standard is still needed to detail the mechanisms.
Autophagy is an essential, homeostatic process by which cells internalize and digest their own components (1414. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469: 323-335, doi: 10.1038/nature09782.
https://doi.org/10.1038/nature09782...
). Although the contribution of autophagy to neuronal death/survival is still under debate (1515. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005; 12 (Suppl 2): 1509-1518, doi: 10.1038/sj.cdd.4401751.
https://doi.org/10.1038/sj.cdd.4401751...
), in many neurodegenerative diseases, such as Alzheimer's disease or Parkinson's disease, autophagy has been found to confer neuroprotection (1616. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13: 805-811, doi: 10.1038/nn.2575.
https://doi.org/10.1038/nn.2575...
). The protective effect of autophagic activation after hypoxic/ischemic injury was initially reported in a C. elegans model (17). 3-Methyladenine (3-MA), an autophagy inhibitor, sensitized the cells to hypoxic injury (1818. Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32: 329-339, doi: 10.1016/j.nbd.2008.07.022.
https://doi.org/10.1016/j.nbd.2008.07.02...
), whereas rapamycin, an autophagy inducer, blocked cell death after ischemic injury (1717. Samokhvalov V, Scott BA, Crowder CM. Autophagy protects against hypoxic injury in C. elegans. Autophagy 2008; 4: 1034-1041.). These data implicate autophagy in the neuroprotection induced by IPC. Moreover, this "self-eating" behavior could partially explain the tolerance involved in IPC protection.
To demonstrate the neuroprotection of IPC on focal ischemic injury and its underlying mechanisms, we applied a series of IPC paradigms in a rat I/R model. Our data clearly addressed the premise that levels of autophagic activation induced by a series of IPC events were correlated with neuroprotection activity after focal ischemic injury. More important, IPC promoted autophagy through mediation of the tuberous sclerosis type 1 (TSC1)-mTor pathway, subsequently attenuating I/R-induced apoptosis. The present study provides a focus for further understanding of the mechanism of endogenous neuroprotection by IPC.
Material and Methods
Focal IPC and permanent middle cerebral artery occlusion
Male Sprague-Dawley rats (weighing 280-300 g) were purchased from the Center for Experimental Animals of Chongqing Medical University (China). The US National Institutes of Health Guidelines for Care and Use of Laboratory Animals were followed in all animal procedures. Focal IPC and/or permanent focal cerebral ischemia (PFI) models were induced by using a suture occlusion technique. Rats were anesthetized by intraperitoneal injection of 4% chloral hydrate (350 mg/kg). Through a ventral midline incision, the common carotid artery on both sides was occluded. Four different IPC paradigms were applied in this study: 5 min (1 episode, 5-min duration), 5×5 min (2 episodes, 5-min duration, 15-min interval), 5×5×5 min (3 episodes, 5-min duration, 15-min intervals), and 15 min (1 episode, 15-min duration). After IPC induction, the suture was gently withdrawn to permit reperfusion. Twenty-four hours later, the suture was reintroduced into the internal carotid through the stump of the external carotid artery to induce PFI (permanent middle cerebral artery occlusion). Sham-operated rats underwent identical surgery, but the suture was not inserted. Laser-Doppler flowmetry (ML191, Laser Doppler Blood Flow Meter, AD Instruments, UK) was used to monitor the blocking of cerebral blood flow. Body temperature was closely monitored with a rectal probe and maintained in the range of 37.0±0.5°C with a heating pad (Institute of Biomedical Engineering, USA, CAMS, BME-412A Animal regulator) during and after surgery until recovery from anesthesia. Rats showing tremors and seizures after surgery were excluded from further experiments.
Evaluation of infarct volume
Twenty-four hours after ischemia injury, the neurological deficits in rats subjected to PFI were evaluated in a blinded manner: 0 points, rats behaved normally; 1 point, rats could not fully stretch their left front legs; 2 points, rats turned around in a circle; 3 points, rats fell down to the left side; 4 points, rats could not move by themselves, losing consciousness. After being scored, the rats were killed, the brains were dissected and sectioned in a plastic module (Harvard Apparatus, USA); 3-mm thick sections were stained with 4% 2,3,5-triphenyltetrazolium chloride (TTC) for 30 min, and fixed with 4% paraformaldehyde. The total wet weight of the TTC-stained brains was quantified with an electronic scale (Mettler-Toledo Group, USA). The wet red and white regions of the TTC-stained brains were collected separately. Infarct volume was analyzed using five 3-mm coronal sections from each brain and calculated using the following formula: infarct volume=(total wet weight−red weight)/total wet weight×100%. After the wet weight of the brains was quantified, the red and white parts of these brains were desiccated at 105°C for 48 h until the weight was constant.
Hematoxylin and eosin (HE) staining
Tissue was rapidly removed from the brain, and washed several times with 0.1 M phosphate-buffered saline (PBS), pH 7.4, fixed in 10% formalin for 24 h at room temperature, dehydrated by graded ethanol, and embedded in paraffin. For HE staining, sections (6-µm thick) were deparaffinized with xylene, stained with HE, and then observed by light microscopy. For analysis, the blue nuclei of four fields from each image were counted.
Immunohistochemistry
The tissue was rapidly removed, washed several times with 0.1 M PBS, pH 7.4, fixed in 10% formalin for 24 h at room temperature, dehydrated by graded ethanol, and embedded in paraffin. Sections (6-µm thick) were deparaffinized with xylene and incubated with the LC3B antibody (monoclonal mouse, 1:500, Cell Signaling Technology, USA ) at 4°C for 24 h. Secondary antibodies were applied for 90 min at room temperature. Slides were developed with diaminobenzidine substrate, and the images were photographed on an Olympus microscope (Olympus Corp., Japan).
TUNEL assay
For the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, slides were developed with diaminobenzidine substrate, counterstained with HE, and then examined for evidence of apoptosis. The number of brown apoptotic cells were normalized to total cells as detected by HE. Four fields of each image were counted.
Western blotting
After IPC and/or PFI, brain tissues from the ischemic cortex and corresponding area of sham-operated rats were homogenized and proteins extracted with a lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton-100, 0.1% SDS, 5 mM EDTA, 1 mM PMSF, 0.28 U/mL aprotinin, 50 µg/mL leupeptin, 1 mM benzamidine, and 7 µg/mL pepstatin A). Protein concentrations were determined (SmartSpec3000 Spectrophotometer, Bio-Rad, USA) using a BCA kit (23227, Pierce, USA). A 60-µg aliquot of proteins from each sample was separated using 10% SDS-PAGE and subsequently transferred to a nitrocellulose membrane. The membranes were then incubated with specific antibodies against LC3 (1:1000; ab62721, Abcam, USA), Beclin (1:500; sc-11427, Santa Cruz Biotechnology, USA), caspase-3 (1:200; sc-24, Santa Cruz Biotechnology, USA), p-mTor or mTor (1:500; Cell Signaling Technology), TSC1 (1:500; Cell Signaling Technology), anti-p-TSC1 Thr417 (1:200; Tebu-Bio, UK), TSC2, or phospho-tuberin/TSC2 (Thr1462) antibody (1:500; Cell Signaling Technology) at 4°C overnight, and incubated with a horseradish peroxidase-conjugated secondary antibody (1:5000; Jackson ImmunoResearch, USA) at room temperature for 1 h. Immunoreactivity was detected by an enhanced chemiluminescent autoradiography (34080, Pierce) in accordance with the manufacturer's instructions. The membranes were reprobed with β-actin (1:5000; A5441, Sigma, USA).
Treatment with autophagy inhibitor or inducer
To determine the effects of pretreatment with the autophagy inhibitor 3-MA on autophagy activation and the neuroprotective effects of IPC, rats were treated with an intracerebral ventricle (icv) injection of 3-MA (300 nM, Sigma) 5 min before the onset of IPC. 3-MA was dissolved in normal saline by heating the solution to 60-70°C immediately before treatment. To explore if rapamycin, an autophagy inducer, could potentiate the effects of IPC, rats were treated with icv injection of rapamycin (20 pM, Sigma) 5 min before the onset of IPC. Rapamycin was first dissolved in ethanol and then diluted to the final concentration with normal saline (final ethanol concentration <2%). Control animals received an icvinjection of the same volume of vehicle.
Statistical analysis
Data are reported as means±SE. Statistical analyses of the data were performed using one-way ANOVA followed by the Bonferroni test, and the Studentt-test. P<0.05 was considered to be a significant difference.
Results
IPC activated autophagy in sham rats and I/R rats
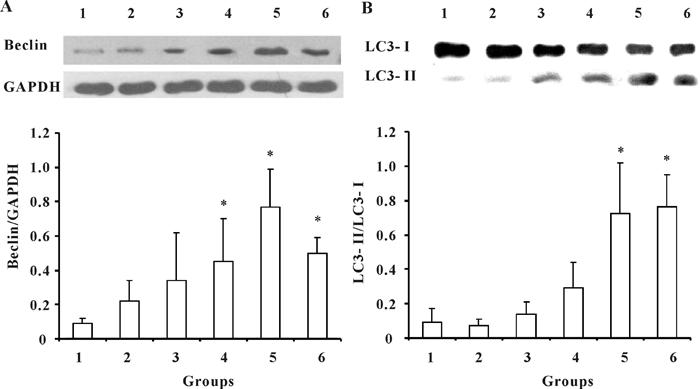
Because IPC possesses the potential to protect against I/R injury, we initially determined whether IPC activated autophagic activity. Four IPC paradigms were applied, and Beclin and LC3, the marker proteins of autophagy, were detected 24 h after IPC. Results showed that IPC increased Beclin expression in an episode-dependent manner (Figure 1A). When compared to the sham-operated group, significant differences were observed in the 5 min, 5×5 min, 5×5×5 min, and 15 min IPC groups, respectively (P<0.05). Although significant enhancement of autophagic activity was observed in the 15 min IPC group in comparison with the sham control group, the activity was much lower than in the 5×5×5 min group. Additionally, autophagic activity was decreased 72 h after PFC by measuring Beclin expression (data not shown). We next detected expression of LC3 protein, the gold standard assay of autophagy. LC3-I/II conversion represents autophagic activation. Consistent with Beclin expression, autophagy activation was further confirmed by LC3-I/II conversion (Figure 1B). More importantly, the 5×5×5 min IPC paradigm induced the highest level of autophagy. Immunohistochemistry data further confirmed that the 5×5×5 IPC group promoted autophagic activity, as LC3B expression was significantly increased (P<0.05; Figure 1C).
Ischemic preconditioning (IPC) activates autophagy in control rats 24 h after surgery. A, IPC increased Beclin expression.B, IPC increased LC3 conversion. Data are reported as means±SE of 4 rats in each group. *P<0.05, compared to sham control group (one-way ANOVA followed by the Bonferroni test). Groups: 1) sham control, 2) 5 min IPC, 3) 5×5 min IPC, 4) 5×5×5 min IPC, and 5) 15 min IPC. C, Representative images of immunohistochemistry of LC3B (bar: 40 µm). Arrows indicate the cytoplasmic expression of LC3B. D, Expression of TSC-mTor pathway related proteins. IPC increased phospho-TSC1 (P-TSC1), while it decreased phospho-mTor (P-mTor) levels in an episode-dependent manner, whereas total mTor, TSC1, TSC2, and P-TSC2 were not affected by IPC. TSC1: tuberous sclerosis type 1; TSC2: tuberous sclerosis type 2.
Because the mTor pathway directly mediates autophagic activity, we detected total mTor, p-mTor, TSC1, p-TSC1, TSC2, and p-TSC2 levels 24 h after IPC. As shown inFigure 1D, IPC increased p-TSC1, while it decreased p-mTor levels in an episode-dependent manner, which was correlated with autophagic activity, whereas total mTor, TSC1, TSC2, and p-TSC2 were not affected by IPC (Figure 1D).
Next, we tested whether IPC altered autophagic activity in I/R rats by detecting Beclin and LC3-I/II conversion again 24 h after I/R. Compared to the sham control, I/R did not increase autophagic activity, as Beclin expression (Figure 2A) and LC3-I/II conversion (Figure 2B) were not affected; however, the 5×5 min, 5×5×5 min, and 15 min IPC groups prior to I/R significantly increased Beclin expression compared to the model group. With regard to LC3-I/II conversion, only the 5×5×5 min and 15 min IPC groups showed significantly increased LC3-I/II conversion when compared to the I/R group. These data indicated that duration, as well as the number of episodes, mediated the extent of autophagic activation, and these factors might together determine the efficacy of IPC in preventing ischemic injury.
Ischemic preconditioning (IPC) activates autophagy in rats undergoing ischemic reperfusion (I/R) 24 h post-ischemic injury.A, IPC increased Beclin expression. B, IPC increased LC3 conversion. Data are reported as means±SE of 4 rats in each group. Groups: 1) sham control, 2) I/R, 3) 5 min IPC+I/R, 4) 5×5 min IPC+I/R, 5) 5×5×5 min IPC+I/R, and 6) 15 min IPC+I/R. *P<0.05, compared to I/R group (one-way ANOVA followed by the Bonferroni test).
IPC attenuates I/R-induced cleaved caspase-3 levels
As reported (1919. Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S, et al. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci U S A 2008; 105: 4892-4897, doi: 10.1073/pnas.0800628105.
https://doi.org/10.1073/pnas.0800628105...
), IPC reduces I/R-induced caspase-3-dependent apoptosis, and we hereby detected that autophagy was protective in ischemic injury. Using both an autophagy inducer and inhibitor, our results showed that the 3×3×3 IPC mode significantly attenuated cleaved caspase-3 expression when compared to the model group (Figure 3). However, 5 minivc pretreated with 3-MA abolished the protection of IPC as active caspase-3 levels were reversed to model levels. By contrast, preapplication of rapamycin, the inducer of autophagy, could potentiate the protection by IPC. As shown in Figure 3, we found that cleaved caspase-3 was further decreased under pretreatment with rapamycin when compared to the IPC group, although the decrease was not significantly different.
Ischemic preconditioning (IPC) inhibits ischemic reperfusion (I/R)-induced cleaved caspase-3 expression 24 h post-ischemic injury. Data are reported as means±SE of 4 rats in each group. Groups: 1) sham control, 2) I/R, 3) 5×5×5 min IPC+I/R, 4) 3-methyladenine+5×5×5 min IPC+I/R, and 5) rapamycin+5×5×5 min IPC+I/R.#P<0.05, compared to sham control group; *P<0.05, compared to I/R group (one-way ANOVA followed by the Bonferroni test).
IPC attenuates I/R-induced apoptosis
IPC increased autophagic activity and decreased I/R-induced active caspase-3. We further used the TUNEL assay to measure apoptosis 24 h after I/R. As shown inFigure 4, a large number of apoptotic cells were observed in the infarcted area of I/R rats, whereas IPC significantly attenuated I/R-induced apoptosis. More important, 3-MA reversed the neuroprotection of IPC against I/R-induced apoptosis, while rapamycin preapplication potentiated neuroprotection. These results indicated that IPC prevented I/R-induced apoptosis through mediation of autophagic activation.
Ischemic preconditioning (IPC) inhibits ischemic reperfusion (I/R)-induced apoptosis 24 h post-ischemic injury (TUNEL staining).A, Representative image of sham control.B, Representative image of I/R. C, Representative image of 5×5×5 min IPC+I/R group.D, Pooled data are reported as means±SE of 4 rats in each group. Groups: 1) sham control, 2) I/R, 3) 5×5×5 min IPC+I/R, 4) 3-methyladenine+5×5×5 min IPC+I/R, and 5) rapamycin+5×5×5 min IPC+I/R. The arrows indicate the apoptotic cells. #P<0.05, compared to sham control group; *P<0.05, compared to I/R group (one-way ANOVA followed by the Bonferroni test).
HE staining showed that I/R caused remarkable cell death in the infarcted area, which was mitigated by IPC. Consistent with the TUNEL assay, 3-MA abolished, whereas rapamycin potentiated, the protection by IPC. These data further confirmed that IPC protected against ischemic insult by inducing autophagy activation (Figure 5).
Ischemic preconditioning (IPC) inhibits ischemic reperfusion (I/R)-induced cell death 24 h post-ischemic injury (HE staining).A, Representative image of sham control.B, Representative image of I/R. C, Representative image of 5×5×5 min IPC + I/R group.D, Pooled data are reported as means±SE of 4 rats in each group. Groups: 1) sham control, 2) I/R, 3) 5×5×5 min IPC+I/R, 4) 3-methyladenine+5×5×5 min IPC+I/R, and 5) rapamycin+5×5×5 min IPC+I/R. Arrows indicate the nucleus staining. #P<0.05, compared to sham control group; *P<0.05, compared to I/R group (one-way ANOVA followed by the Bonferroni test).
IPC attenuates I/R-induced neurological injury and infarction volume
We confirmed the protective activity of IPC by measuring neurological scores (Figure 6A) and infarct volume (Figure 6B) after ischemic insult. Our data showed that the 5×5×5 min paradigm provided the optimal protection from I/R injury. Additionally, 3-MA abolished, whereas rapamycin potentiated, the protective effect of IPC.
Ischemic preconditioning (IPC) protects against ischemic reperfusion (I/R)-induced neurological damage 24 h post-ischemic injury.A, IPC attenuates I/R-induced increase in neurological score. B, IPC attenuates I/R-induced increase in infarct volume. Data are reported as means±SE of 4 rats in each group. Groups: 1) sham control, 2) I/R, 3) 5×5×5 min IPC+I/R, 4) 3-methyladenine+5×5×5 min IPC+I/R, and 5) rapamycin+5×5×5 min IPC+I/R.#P<0.05, compared to sham control group; *P<0.05, compared to I/R group (one-way ANOVA followed by the Bonferroni test).C, Scheme illustrates the possible protective mechanisms of IPC against ischemic injury through tuberous sclerosis type 1 (TSC1)-mTor-mediated autophagic activation, which inhibited apoptosis induced by ischemic perfusion.
Discussion
Many reports have demonstrated that IPC protects against ischemic injury (1111. Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29: 1937-1950, doi: 10.1161/01.STR.29.9.1937.
https://doi.org/10.1161/01.STR.29.9.1937...
12. Zimmerman RF, Ezeanuna PU, Kane JC, Cleland CD, Kempananjappa TJ, Lucas FL, et al. Ischemic preconditioning at a remote site prevents acute kidney injury in patients following cardiac surgery. Kidney Int 2011; 80: 861-867, doi: 10.1038/ki.2011.156.
https://doi.org/10.1038/ki.2011.156...
-1313. Quarrie R, Cramer BM, Lee DS, Steinbaugh GE, Erdahl W, Pfeiffer DR, et al. Ischemic preconditioning decreases mitochondrial proton leak and reactive oxygen species production in the postischemic heart. J Surg Res 2011; 165: 5-14, doi: 10.1016/j.jss.2010.09.018.
https://doi.org/10.1016/j.jss.2010.09.01...
). Interestingly, IPC-related stratagies have been used to treat ischemic injury clinically (2020. Kloner RA. Clinical application of remote ischemic preconditioning. Circulation 2009; 119: 776-778, doi: 10.1161/CIRCULATIONAHA.108.832832.
https://doi.org/10.1161/CIRCULATIONAHA.1...
, 2121. Cheung MM, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, et al. Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans. J Am Coll Cardiol 2006; 47: 2277-2282, doi: 10.1016/j.jacc.2006.01.066.
https://doi.org/10.1016/j.jacc.2006.01.0...
). Either local or remote IPC can be applied to reduce injury to an organ or tissue (2222. Dave KR, Saul I, Prado R, Busto R, Perez-Pinzon MA. Remote organ ischemic preconditioning protect brain from ischemic damage following asphyxial cardiac arrest. Neurosci Lett 2006; 404: 170-175, doi: 10.1016/j.neulet.2006.05.037.
https://doi.org/10.1016/j.neulet.2006.05...
). For example, the preconditioning stimulus through suprasystolic blood pressure inflation on an arm or leg will confer myocardial protection against subsequent ischemia (2020. Kloner RA. Clinical application of remote ischemic preconditioning. Circulation 2009; 119: 776-778, doi: 10.1161/CIRCULATIONAHA.108.832832.
https://doi.org/10.1161/CIRCULATIONAHA.1...
). Transient ischemic attacks before ischemic stroke could decrease the lesion size as evidenced by a multicenter magnetic resonance imaging study (88. Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, et al. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke 2004; 35: 616-621, doi: 10.1161/01.STR.0000115767.17923.6A.
https://doi.org/10.1161/01.STR.000011576...
, 2323. Johnston SC. Ischemic preconditioning from transient ischemic attacks? Data from the Northern California TIA Study. Stroke 2004; 35: 2680-2682, doi: 10.1161/01.STR.0000143322.20491.0f.
https://doi.org/10.1161/01.STR.000014332...
). Nevertheless, the exact mechanisms underlying the tolerance of ischemic insult remain elusive. In this study, by applying different IPC paradigms, we determined that 5×5×5 min IPC was the most effective in protecting against cerebral I/R injury.
IPC was first described in 1986 by Murry et al. (2424. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124-1136, doi: 10.1161/01.CIR.74.5.1124.
https://doi.org/10.1161/01.CIR.74.5.1124...
) using a dog model; brief episodes of ischemia (4 cycles of 5-min occlusion followed by reperfusion) reduced the extent of infarction induced by subsequent prolonged occlusion. Despite the length of time that has elapsed since the discovery of IPC, a detailed exposition of its mechanism and evidence of the biological processes are still unknown. Autophagy is one of the most convincing mechanisms to explain tolerance of or adjustment to ischemia. In our initial study, we found that IPC induced autophagic activation lasting for at least 72 h. In this study, 3-MA abolished, while rapamycin potentiated, the neuroprotection afforded by IPC. Overall, the data clearly demonstrated that autophagy activation induced by IPC was neuroprotective against ischemic insult.
Sublethal stress activated autophagy but did not activate a lethal ischemic insult, which is consistent with previous reports (2525. Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010; 6: 482-494, doi: 10.4161/auto.6.4.11737.
https://doi.org/10.4161/auto.6.4.11737...
). Although some reports indicated that lethal ischemic insult also activated autophagy, it was suggested that autophagic activation in the ischemic process was neuroprotection-related. By contrast, autophagic activation involved in perfusion was likely neurodetrimental (2626. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100: 914-922, doi: 10.1161/01.RES.0000261924.76669.36.
https://doi.org/10.1161/01.RES.000026192...
). Similarly, treatment with chloroquine resulted in accumulation of LC3-II in sham-operated rats, but did not change the LC3-II levels in postischemic brain tissues (2727. Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem 2010; 115: 68-78, doi: 10.1111/j.1471-4159.2010.06905.x.
https://doi.org/10.1111/j.1471-4159.2010...
). These results indicate that at least part of the accumulation of protein aggregate-associated organelles seen following ischemia is likely due to failure of the autophagy pathway.
Meanwhile, we demonstrated that IPC decreased p-mTor levels (Figure 1D). Since mTor negatively mediates autophagy, this decrease might well explain the IPC-induced autophagic activation. Upstream of mTORC1 is the TSC1-TSC2 inhibitory complex, which functions as a GTPase-activating protein for the GTPase Rheb, an upstream activator of mTor. The TSC1-TSC2 complex inactivates Rheb to inhibit mTOR signaling, which would lead to autophagy. Indeed, we found that IPC increased p-TSC1 levels, while it did not affect p-TSC2, total TSC1, and total TSC2. Nevertheless, this detailed signal pathway deserves further study.
We further demonstrated that cerebral I/R caused cell death through inducing capase-3-dependent apoptosis. We clearly showed apoptotic cell death in the I/R model, but not in IPC rats. Importantly, IPC prevented I/R-induced apoptosis. All these data were consistent with the infarction volume and neurological protection observed in the different groups. In contrast to the neuroprotection of autophagy, apoptosis played a role in cell death that occurred in I/R. Therefore, apoptosis should be the therapeutic target in ischemic injury treatment, while autophagy could be proposed as an important intervening inducer. With regard to the apoptotic mechanisms involved in ischemic reperfusion, both caspase-dependent and caspase-independent pathways have been reported (2828. Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci U S A 1999; 96: 5752-5757, doi: 10.1073/pnas.96.10.5752.
https://doi.org/10.1073/pnas.96.10.5752...
, 2929. Cho BB, Toledo-Pereyra LH. Caspase-independent programmed cell death following ischemic stroke. J Invest Surg 2008; 21: 141-147, doi: 10.1080/08941930802029945.
https://doi.org/10.1080/0894193080202994...
). In the present study, we demonstrated that caspase-3 expression was upregulated by ischemic injury and attenuated by IPC.
In conclusion, the present study suggests that three episodes of IPC protected against I/R-induced apoptosis through autophagy activation, with subsequent neuroprotection (Figure 6C). This study provides further experimental evidence for the protection of IPC against ischemic injury and the application of IPC in stroke therapy.
References
-
1Hankey GJ, Jamrozik K, Broadhurst RJ, Forbes S, Burvill PW, Anderson CS, et al. Five-year survival after first-ever stroke and related prognostic factors in the Perth Community Stroke Study. Stroke 2000; 31: 2080-2086, doi: 10.1161/01.STR.31.9.2080.
» https://doi.org/10.1161/01.STR.31.9.2080 -
2Gosman-Hedstrom G, Claesson L, Klingenstierna U, Carlsson J, Olausson B, Frizell M, et al. Effects of acupuncture treatment on daily life activities and quality of life: a controlled, prospective, and randomized study of acute stroke patients. Stroke 1998; 29: 2100-2108, doi: 10.1161/01.STR.29.10.2100.
» https://doi.org/10.1161/01.STR.29.10.2100 -
3Naeser MA, Hamblin MR. Potential for transcranial laser or LED therapy to treat stroke, traumatic brain injury, and neurodegenerative disease. Photomed Laser Surg 2011; 29: 443-446, doi: 10.1089/pho.2011.9908.
» https://doi.org/10.1089/pho.2011.9908 -
4Minnerup J, Schabitz WR. Improving outcome after stroke: time to treat new targets. Stroke 2012; 43: 295-296, doi: 10.1161/STROKEAHA.111.642363.
» https://doi.org/10.1161/STROKEAHA.111.642363 -
5Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 2012; 79: 1853-1861, doi: 10.1212/WNL.0b013e318271f76a.
» https://doi.org/10.1212/WNL.0b013e318271f76a -
6Pignataro G, Scorziello A, Di RG, Annunziato L. Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J 2009; 276: 46-57, doi: 10.1111/j.1742-4658.2008.06769.x.
» https://doi.org/10.1111/j.1742-4658.2008.06769.x -
7Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, et al. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke 2004; 35: 2687-2689, doi: 10.1161/01.STR.0000143216.85349.9e.
» https://doi.org/10.1161/01.STR.0000143216.85349.9e -
8Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, et al. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke 2004; 35: 616-621, doi: 10.1161/01.STR.0000115767.17923.6A.
» https://doi.org/10.1161/01.STR.0000115767.17923.6A -
9Schaller B, Graf R. Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol 2002; 249: 1503-1511, doi: 10.1007/s00415-002-0933-8.
» https://doi.org/10.1007/s00415-002-0933-8 -
10Janoff A, Weissmann G, Zweiofach BW, Thomas L. Pathogenesis of experimental shock. IV. Studies on lysosomes in normal and tolerant animals subjected to lethal trauma and endotoxemia. J Exp Med 1962; 116: 451-466, doi: 10.1084/jem.116.4.451.
» https://doi.org/10.1084/jem.116.4.451 -
11Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29: 1937-1950, doi: 10.1161/01.STR.29.9.1937.
» https://doi.org/10.1161/01.STR.29.9.1937 -
12Zimmerman RF, Ezeanuna PU, Kane JC, Cleland CD, Kempananjappa TJ, Lucas FL, et al. Ischemic preconditioning at a remote site prevents acute kidney injury in patients following cardiac surgery. Kidney Int 2011; 80: 861-867, doi: 10.1038/ki.2011.156.
» https://doi.org/10.1038/ki.2011.156 -
13Quarrie R, Cramer BM, Lee DS, Steinbaugh GE, Erdahl W, Pfeiffer DR, et al. Ischemic preconditioning decreases mitochondrial proton leak and reactive oxygen species production in the postischemic heart. J Surg Res 2011; 165: 5-14, doi: 10.1016/j.jss.2010.09.018.
» https://doi.org/10.1016/j.jss.2010.09.018 -
14Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469: 323-335, doi: 10.1038/nature09782.
» https://doi.org/10.1038/nature09782 -
15Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005; 12 (Suppl 2): 1509-1518, doi: 10.1038/sj.cdd.4401751.
» https://doi.org/10.1038/sj.cdd.4401751 -
16Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13: 805-811, doi: 10.1038/nn.2575.
» https://doi.org/10.1038/nn.2575 -
17Samokhvalov V, Scott BA, Crowder CM. Autophagy protects against hypoxic injury in C. elegans Autophagy 2008; 4: 1034-1041.
-
18Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32: 329-339, doi: 10.1016/j.nbd.2008.07.022.
» https://doi.org/10.1016/j.nbd.2008.07.022 -
19Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S, et al. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci U S A 2008; 105: 4892-4897, doi: 10.1073/pnas.0800628105.
» https://doi.org/10.1073/pnas.0800628105 -
20Kloner RA. Clinical application of remote ischemic preconditioning. Circulation 2009; 119: 776-778, doi: 10.1161/CIRCULATIONAHA.108.832832.
» https://doi.org/10.1161/CIRCULATIONAHA.108.832832 -
21Cheung MM, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, et al. Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans. J Am Coll Cardiol 2006; 47: 2277-2282, doi: 10.1016/j.jacc.2006.01.066.
» https://doi.org/10.1016/j.jacc.2006.01.066 -
22Dave KR, Saul I, Prado R, Busto R, Perez-Pinzon MA. Remote organ ischemic preconditioning protect brain from ischemic damage following asphyxial cardiac arrest. Neurosci Lett 2006; 404: 170-175, doi: 10.1016/j.neulet.2006.05.037.
» https://doi.org/10.1016/j.neulet.2006.05.037 -
23Johnston SC. Ischemic preconditioning from transient ischemic attacks? Data from the Northern California TIA Study. Stroke 2004; 35: 2680-2682, doi: 10.1161/01.STR.0000143322.20491.0f.
» https://doi.org/10.1161/01.STR.0000143322.20491.0f -
24Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124-1136, doi: 10.1161/01.CIR.74.5.1124.
» https://doi.org/10.1161/01.CIR.74.5.1124 -
25Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010; 6: 482-494, doi: 10.4161/auto.6.4.11737.
» https://doi.org/10.4161/auto.6.4.11737 -
26Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100: 914-922, doi: 10.1161/01.RES.0000261924.76669.36.
» https://doi.org/10.1161/01.RES.0000261924.76669.36 -
27Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem 2010; 115: 68-78, doi: 10.1111/j.1471-4159.2010.06905.x.
» https://doi.org/10.1111/j.1471-4159.2010.06905.x -
28Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci U S A 1999; 96: 5752-5757, doi: 10.1073/pnas.96.10.5752.
» https://doi.org/10.1073/pnas.96.10.5752 -
29Cho BB, Toledo-Pereyra LH. Caspase-independent programmed cell death following ischemic stroke. J Invest Surg 2008; 21: 141-147, doi: 10.1080/08941930802029945.
» https://doi.org/10.1080/08941930802029945
-
First published online July 16, 2013.
Publication Dates
-
Publication in this collection
16 July 2013 -
Date of issue
Aug 2013
History
-
Received
1 Dec 2012 -
Accepted
3 May 2013