Resumos
Considerando a composição da multimistura distribuída em Belo Horizonte/MG (trigo, fubá, casca de ovo e folha de mandioca), as metodologias de detecção e/ou quantificação dos fitatos e oxalatos foram validadas. A purificação em coluna de troca aniônica forte do ácido fítico e seus derivados desfosforilados extraídos da multimistura apresentou baixos valores de recuperação (49%) nas concentrações menores do fitato adicionado, em relação às mais elevadas (101%), sugerindo a interferência de minerais presentes em altas concentrações, principalmente cálcio. Dos métodos avaliados para a determinação do ácido oxálico, a cromatografia de exclusão iônica foi a que apresentou os melhores resultados, com boas recuperações nos níveis mais altos de adição (88%), apresentando, contudo, recuperação mais baixa nos níveis inferiores de adição (61%). Os teores de ácido fítico nas amostras variaram de 1,61 g/100 g a 2,25 g/100 g e, para o ácido oxálico, de 0,044 g/100 g a 0,057 g/100 g. Os critérios de validação dos métodos analíticos por cromatografia líquida de alta eficiência foram considerados satisfatórios quanto a seletividade e especificidade, linearidade, repetitividade e limites de detecção e quantificação. Contudo, o de recuperação não foi atendido satisfatoriamente, visto que as porcentagens obtidas foram inferiores ao esperado, levando-se em conta a concentração adicionada nas amostras.
ácido fítico; acido oxálico; cromatografia; fitatos; complemento alimentar; validação
Taking into account the composition of the "multimistura" distributed in Belo Horizonte/MG (wheat, corn meal, egg shell and cassava leave), the methods of detection and/or quantitation of phytates and oxalates were validated. The purification of phytic acid and its dephosphorylated derivatives extracted from the "multimistura" using a strong anionic exchange column presented low values of recovery (49%) at low concentrations of the added phytate, when related to the higher ones (101%), which suggests interference of minerals present in high levels, mainly calcium. From the methods evaluated for the determination of oxalic acid, the ionic exclusion chromatography was the one that presented the best results, with good recoveries at the highest addition levels (88%); presenting, however, decreased recovery in the lower levels of addition (61%). The levels of phytic acid in the samples varied from 1.61 g/100 g to 2.25 g/100 g and for the oxalic acid, from 0.044 g/100 g to 0.057 g/100 g. The validation criteria for the analytical methods by high performance liquid chromatography were considered satisfactory for selectivity and specificity, linearity, repetitivity and limits of detection and quantitation. However, the recovery was not satisfactory since the obtained percentages were lower than the expected one, taking into account the quantities added to the samples.
oxalic acid; phytic acid; chromatography; phytates; food complement; validation
Validação de métodos para determinação dos ácidos fítico e oxálico em multimistura
Validation methods to determine phytic and oxalic acids in "multimisturas"
Giancarlo Ubaldo NappiI; Mariem Rodrigues Ribeiro-CunhaI; José Virgílio CoelhoII; Lieselotte JoklII, * * A quem a correspondência deve ser enviada
IServiço de Química, Divisão de Vigilância Sanitária, Instituto Octávio Magalhães, Fundação Ezequiel Dias
IIFaculdade de Farmácia da Universidade Federal de Minas Gerais, R. Gal Andrade Neves, 76/4, CEP 30430-070, Belo Horizonte (MG), Brasil, E-mail: lisejokl@ufmg.br
RESUMO
Considerando a composição da multimistura distribuída em Belo Horizonte/MG (trigo, fubá, casca de ovo e folha de mandioca), as metodologias de detecção e/ou quantificação dos fitatos e oxalatos foram validadas. A purificação em coluna de troca aniônica forte do ácido fítico e seus derivados desfosforilados extraídos da multimistura apresentou baixos valores de recuperação (49%) nas concentrações menores do fitato adicionado, em relação às mais elevadas (101%), sugerindo a interferência de minerais presentes em altas concentrações, principalmente cálcio. Dos métodos avaliados para a determinação do ácido oxálico, a cromatografia de exclusão iônica foi a que apresentou os melhores resultados, com boas recuperações nos níveis mais altos de adição (88%), apresentando, contudo, recuperação mais baixa nos níveis inferiores de adição (61%). Os teores de ácido fítico nas amostras variaram de 1,61 g/100 g a 2,25 g/100 g e, para o ácido oxálico, de 0,044 g/100 g a 0,057 g/100 g. Os critérios de validação dos métodos analíticos por cromatografia líquida de alta eficiência foram considerados satisfatórios quanto a seletividade e especificidade, linearidade, repetitividade e limites de detecção e quantificação. Contudo, o de recuperação não foi atendido satisfatoriamente, visto que as porcentagens obtidas foram inferiores ao esperado, levando-se em conta a concentração adicionada nas amostras.
Palavras-chave: ácido fítico, acido oxálico, cromatografia, fitatos, complemento alimentar, validação.
SUMMARY
Taking into account the composition of the "multimistura" distributed in Belo Horizonte/MG (wheat, corn meal, egg shell and cassava leave), the methods of detection and/or quantitation of phytates and oxalates were validated. The purification of phytic acid and its dephosphorylated derivatives extracted from the "multimistura" using a strong anionic exchange column presented low values of recovery (49%) at low concentrations of the added phytate, when related to the higher ones (101%), which suggests interference of minerals present in high levels, mainly calcium. From the methods evaluated for the determination of oxalic acid, the ionic exclusion chromatography was the one that presented the best results, with good recoveries at the highest addition levels (88%); presenting, however, decreased recovery in the lower levels of addition (61%). The levels of phytic acid in the samples varied from 1.61 g/100 g to 2.25 g/100 g and for the oxalic acid, from 0.044 g/100 g to 0.057 g/100 g. The validation criteria for the analytical methods by high performance liquid chromatography were considered satisfactory for selectivity and specificity, linearity, repetitivity and limits of detection and quantitation. However, the recovery was not satisfactory since the obtained percentages were lower than the expected one, taking into account the quantities added to the samples.
Keywords: oxalic acid, phytic acid, chromatography, phytates, food complement, validation.
1 - INTRODUÇÃO
A multimistura é um complemento alimentar de baixo custo e de fácil utilização. Nela se aproveita toda a potencialidade nutritiva de alimentos, como folhas de hortaliças, grãos e farelos. Os últimos são subprodutos do beneficiamento de cereais (arroz e trigo), tradicionalmente usados no Brasil para extração de óleos e na nutrição animal. Empregam-se em decorrência das práticas culinárias tradicionais e regionais os brotos de chuchu, folhas e talos de brócolis e casca de ovos, entre outras fontes [4, 11, 46]. Tendo em vista que estes alimentos alternativos são constituídos, basicamente, de fontes de origem vegetal, deve-se levar em consideração, também, a presença de grande variedade de substâncias químicas conhecidas por exercerem efeitos deletérios quando ingeridas pelo homem, como os fitatos e os oxalatos [8, 13, 29, 41, 46].
Assim, levando-se em conta que a multimistura fornecida pela Prefeitura de Belo Horizonte como complemento alimentar a crianças, gestantes e nutrizes do Projeto de Prevenção e Combate à Desnutrição é constituída de 33% de farinha de trigo, 33% de farelo de trigo torrado, 32% de fubá torrado, 1% de pó de casca de ovo e 1% de pó de folha de mandioca, considerou-se que alguns fatores antinutricionais seriam passíveis de estarem presentes em quantidades significativas nesta multimistura: o fitato (hexafosfato de inositol IP6) e seus derivados desfosforilados, encontrados, principalmente, nos farelos e o ácido oxálico, no pó de folhas de mandioca. Na determinação destes compostos, é necessário que os métodos analíticos estejam devidamente validados para que os valores encontrados sejam dignos de confiança e possam estar de acordo com a legislação vigente [5].
A maioria dos métodos analíticos para determinar o ácido fítico se baseia em seu elevado grau de quelação com o íon férrico, formando um complexo insolúvel, em meio ácido [36]. Tais métodos envolvem a extração ácida da amostra e subseqüente precipitação do complexo Fe(III)-fitato formado pela adição de cloreto férrico. Entretanto, estes métodos são pouco sensíveis devido à falta de especificidade para distinguir entre o IP6 e seus isômeros parcialmente desfosforilados que precipitam junto com o ácido fítico [49].
Outro método utilizado é o da AOAC [2], desenvolvido por HARLAND & OBERLEAS [16]. Este se baseia na extração ácida da amostra seguida de purificação em coluna de troca iônica, mediante a eluição do ácido fítico com cloreto de sódio 0,7 mol/L (gradiente não-linear) e digestão do eluato até fosfato inorgânico. Contudo, segundo LEHRFELD & MORRIS [25], este método pode levar à superestimativa do ácido fítico, uma vez que inclui os outros fosfatos de inositol (penta, tetra, tri, bi e mono), mas não os distingue.
A partir da década de 1980, a cromatografia líquida de alta eficiência (CLAE) começou a ser empregada como método analítico para determinar o ácido fítico e seus derivados. O procedimento se baseia na extração ácida das amostras seguida de purificação em coluna de troca aniônica forte (SAX), secagem do eluato até resíduo e dissolução do resíduo na fase móvel antes da injeção. O método de detecção mais freqüentemente utilizado é o índice de refração embora a espectrofotometria de absorção atômica também tenha sido usada [9]. Neste caso, a análise de complexos metal-fitato foi executada acoplando-se o cromatógrafo líquido a um espectrômetro de absorção atômica junto à saída do detector de índice de refração e utilizando como padrão fitato monoférrico.
Desde as primeiras pesquisas baseadas na cromatografia de fase reversa [9, 22, 23], foi demonstrada a eficiência da coluna aniônica nos processos de purificação e concentração das amostras. LEHRFELD [24] e SANDBERG et al. [40] já haviam demonstrado o uso de par iônico na fase reversa para identificar e quantificar este composto e seus derivados desfosforilados, concluindo que suas separações e quantificações foram eficientes. MAYR [30] e ROUNDS & NIELSEN [38] desenvolveram métodos de análise por cromatografia líquida por troca iônica que, apesar de serem mais sensíveis e seletivos que o de fase reversa [6], apresentam o inconveniente de utilizar reação pós-coluna, o que torna o método mais caro e complexo.
Os procedimentos de CLAE, embora bastante sensíveis e seletivos, ainda têm como inconveniente o fato de serem muito demorados e caros. Contudo, são os procedimentos de escolha pelo fato de permitirem a identificação e quantificação do ácido fítico e seus derivados desfosforilados, seja por adição de padrões na cromatografia de fase reversa, seja por reações pós-coluna na cromatografia de troca iônica.
Até o ano de 1977, os procedimentos mais comuns para a determinação de ácido oxálico eram a titulação por permanganato, alguns métodos colorimétricos e a cromatografia por papel. Estes métodos envolvem, basicamente, os mesmos passos: extração do ácido oxálico da matriz vegetal com ácido clorídrico ou carbonato de sódio e precipitação do oxalato de cálcio, seguido pelo tratamento do precipitado com ácido sulfúrico diluído para formar solução de ácido oxálico. O ácido oxálico é quantificado por um dos seguintes métodos: potenciometria com eletrodo de quinidrona [35], titulação com solução padronizada de permanganato de potássio [3] ou colorimetricamente pela conversão do ácido oxálico a ácido glicólico e derivação com o ácido 2,7-dissulfônico-3,6-diidroxinaftaleno [51]. Nestes métodos, foram encontrados resultados conflitantes, principalmente quanto à completa extração do ácido.
Outros métodos mais precisos, como a cromatografia gasosa e uso de radioisótopos eram utilizados, mas em pequena escala [18]. Posteriormente, com o desenvolvimento de técnicas enzimáticas [34], de cromatografia gasosa [42] e de cromatografia líquida [7, 27, 48], estas se tornaram as técnicas de escolha, sempre que possível.
A determinação de ácido oxálico em alimentos foi estudada por ALENCAR & SILVESTRE [1] que compararam três métodos diferentes da literatura. Destes, o de FRANCO & KRINITZ [12] foi o que apresentou melhor recuperação e reprodutibilidade. Entretanto, este método requer quantidade elevada de amostra (aproximadamente 35 g), que nem sempre se tem à disposição. LOURES & JOKL [28] padronizaram o método para pequenas quantidades de amostras, mantendo, ao mesmo tempo, seu elevado nível de recuperação e de reprodutibilidade.
A cromatografia gasosa tem sido usada na determinação de ácido oxálico em alimentos [14], contudo, esta técnica tem a desvantagem de ser necessário esterificar o ácido oxálico antes da análise, aumentando-se assim o número de etapas necessárias para sua determinação.
A CLAE é outra técnica utilizada. Pode ser realizada tanto em fase reversa [26, 27, 48], como por exclusão iônica [37, 43, 44, 47, 50]. Como variante desta última, tem-se o emprego do método da reação enzimática (oxalato oxidase) e detecção amperométrica [19], que apesar de ser mais sensível e seletivo que os procedimentos anteriores, apresenta o inconveniente de executar reação pós-coluna, o que torna o método mais complexo e caro.
Deste modo, os objetivos deste trabalho foram validar metodologias analíticas para determinar e quantificar o ácido fítico e seus derivados e o ácido oxálico. Mais especificamente pretendeu-se:
avaliar a eficiência do método de purificação em coluna de troca aniônica forte para ácido fítico e seus derivados desfosforilados;
validar as condições analíticas para determinar e quantificar o ácido fítico e seus isômeros por CLAE, usando detector de índice de refração;
aplicar as metodologias selecionadas nas respectivas análises destes antinutrientes em multimistura.
2 - MATERIAL E MÉTODOS
2.1 - Amostras
As amostras de multimistura foram fornecidas pela Indústria Prolácteos, sediada no município de Contagem, Minas Gerais, para a Secretaria Municipal de Abastecimento. Foram repassadas mensalmente à Fundação Ezequiel Dias, na forma de cinco unidades de 1 quilograma cada, do mesmo lote de produção, por um período de 10 meses: de outubro de 2002 a junho de 2003. Na seleção das amostras para análise [31, 45], foram escolhidas aleatoriamente 3 amostras de cada 5 fornecidas, a cada mês, perfazendo um total de 30 amostras analisadas.
As amostras selecionadas foram passadas em moinho com malha de 1 mm e preparadas para doseamento do ácido fítico e seus derivados [10, 15] e do ácido oxálico [28].
2.2 - Equipamentos e padrões
Foi utilizado o cromatógrafo líquido de alta eficiência (Shimadzu, Kyoto Japan), composto de bomba LC-10AD; injetor automático SIL-10A com capacidade máxima de injeção de 50 µL; forno para coluna CTO-10A; unidade de desgaseificação DGU-2A. Além disto, usou-se balança analítica, com precisão de 0,05 mg; sistema Millipore® para filtração a vácuo; bomba de vácuo seca Edwards®, modelo A480-01-880; sistema de purificação de água MILLI-Q®, modelo ELIX 3; evaporador centrifugal e agitadores planetário e do tipo vórtex.
Para o ácido fítico e seus derivados empregou-se o detector de índice de refração RID-6A e as colunas de troca aniônica forte (amina aromática ligada à sílica) Lichrolut® SAX 500 mg (3 mL), conectada a um sistema de vácuo e de fase reversa Spherisorb® ODS C18 (5 µm), 250 mm x 4 mm. Enquanto para o ácido oxálico, usaram-se os seguintes detectores: o espectrofotométrico UV/VIS SPD-10A e o de arranjo de diodo (DAD) SPD-M10A e as colunas cromatográficas Lichrospher® 100 RP18 (5 µm), 250 mm x 4,6 mm e 100 RP8® (5 µm), 250 mm x 4,6 mm; e de exclusão iônica Supelcogel® H, 300 mm x 7,8 mm.
Como reagentes, foram utilizados os seguintes padrões da SIGMA: sais do hexafosfato de inositol (IP6 cód. P3168/lote 76H1168), pentafosfato de inositol (IP5 cód. I-9261/lote 28H03591), 1,3,4,5 tetrafosfato de inositol (IP4 cód. I-0143/lote 27H01461) e 1,4,5 trifosfato de inositol (IP3 cód. I-8761/lote 21H84341) e da MERCK: ácido oxálico (cód. 1.00495.1000/lote B252695 823).
2.3 - Determinação da umidade
Imediatamente após terem sido abertos os pacotes selecionados para as análises de ácidos fítico e oxálico, foram retiradas alíquotas em duplicata e pesados 3 g, para determinação de perdas por dessecação em estufa a 100 a 105 °C [21]. Os valores obtidos foram utilizados nos cálculos dos teores de ácido fítico e oxálico, na base seca.
2.4 - Determinação do ácido fítico e seus derivados
2.4.1 - Extração
A extração foi realizada de acordo com SANDBERG & AHDERINNE [39] com algumas modificações introduzidas por CUADRADO et al. [10]. Foram pesadas em duplicata 0,5 g das amostras de cada pacote em tubos de centrífuga de 100 mL de capacidade. A seguir, foram adicionados 20 mL de ácido clorídrico 0,5 mol/L e os conteúdos submetidos à agitação mecânica por 2 h. Após este período, as amostras foram centrifugadas a 289 g por 20 min e os sobrenadantes filtrados em papel quantitativo lento e, a seguir, em filtro de 0,45 micra. Dos filtrados foram pipetados 10 mL, transferidos para tubos de centrífuga de 15 mL de capacidade e evaporados até resíduo. Aos resíduos foram adicionados 15 mL de ácido clorídrico 25 mmol/L.
2.4.2 - Purificação
A purificação das amostras foi realizada de acordo com SANDBERG & AHDERINNE [39] com algumas modificações introduzidas por CUADRADO et al. [10], empregando-se coluna SAX. A coluna foi lavada sucessivamente com 10 e 5 mL de HCl 0,025 mol/L e os fosfatidilinositóis retidos na coluna foram eluídos com 10 mL de HCl 2 mol/L. Os eluatos foram concentrados até resíduo sólido e retomados em 1,0 mL de fase móvel e submetidos às análises quantitativas por CLAE.
2.4.3 - Quantificação por CLAE
A análise por CLAE em fase reversa foi realizada de acordo com CUADRADO et al. [10] com modificação. Na fase móvel, usou-se o tampão composto de 900 mL de metanol + 900 mL de ácido fórmico 0,05 mol/L + 45 mL de EDTA 0,005 mol/L + 54 mL de hidróxido de tetrabutilamônio a 20 mL/100 mL + 1,8 mL da solução de ácido fítico a 0,6 g/100 mL (previamente hidrolisada em autoclave a 127 °C, por 30 min) e o pH ajustado para 4,3 com ácido sulfúrico 9 mol/L. O fitato hidrolisado foi adicionado à fase para garantir que os íons metálicos presentes no sistema (tubulação, coluna e possíveis contaminantes da fase móvel) não quelem os fitatos presentes na amostra. Como fase estacionária foi utilizada a coluna de fase reversa C18, termoestatizada a 45 °C por 1 h e vazão de 1,2 mL/min. O volume injetado foi de 20 µL. Foi usado o detector de índice de refração para determinar o ácido fítico e seus derivados desfosforilados, mediante análise cromatográfica do branco, dos padrões, da amostra adicionada de IP6, da simetria dos picos e da concordância dos tempos de retenção.
2.5 - Determinação do ácido oxálico
2.5.1 - Extração
O ácido oxálico foi extraído [27] após a homogeneização e pesagem de cerca de 5 g da amostra. Para isto, foram adicionados 200 mL de ácido clorídrico 1 mol/L, a extração foi realizada em banho-maria a 100 °C por 15 min e, em seguida, os recipientes, mantidos à temperatura ambiente por uma noite. O extrato foi filtrado em papel de filtro quantitativo lento e diluído com água na proporção 1:1. Antes da injeção, foram passados 4 mL desta solução por cartucho Sep-pak® C18 de 500 mg previamente ativado com 2 mL de metanol e 2 mL de ácido clorídrico 0,5 mol/L.
2.5.2 - Quantificação por CLAE
As amostras injetadas na coluna de exclusão iônica foram preparadas conforme recomendado por LIBERT [27]. Contudo, antes da injeção, o filtrado foi ajustado de forma a se obter um pH próximo ao da fase móvel com fosfato de sódio tribásico 20 g/100 mL. A seguir, as soluções resultantes foram homogeneizadas por 20 segundos em agitador tipo vórtex e filtradas em filtro de 0,45 micra. Foi empregada a coluna de exclusão iônica termoestatizada a 40 °C por 1 h, usando-se como fase móvel ácido ortofosfórico 0,1 g/100 mL. A vazão foi de 0,5 mL/min para o volume injetado de 20 µL. A determinação foi realizada com detector UV/VIS ajustado para leitura no comprimento de onda de 220 nm e o ácido oxálico foi identificado pelo seu espectro na região do ultravioleta por meio do DAD.
2.6 - Validação das metodologias e avaliação dos critérios de desempenho
Os métodos foram validados levando-se em conta os seguintes parâmetros: seletividade e especificidade, linearidade, repetitividade, recuperação e limites de detecção e de quantificação [20].
2.6.1 - Seletividade e especificidade
Tanto a seletividade como a especificidade foram determinadas analisando-se cromatograficamente e individualmente o branco da amostra, o branco dos reagentes, a amostra adicionada dos padrões de IP6 e seus derivados ou ácido oxálico e os padrões propriamente ditos, para se verificar a ausência de picos interferentes nos respectivos cromatogramas.
2.6.2 - Linearidade
A linearidade foi determinada por meio da curva de calibração estabelecida com 7 concentrações (abcissa) e as áreas dos picos (ordenada). A solução de cada concentração foi injetada em triplicata e foram determinados os coeficientes angular e linear da reta de regressão. A seguir, foi aplicado o teste estatístico para a determinação da linearidade para 7 concentrações com p < 0,05. Foram determinados, em seguida, os intervalos de confiança dos valores dos coeficientes angular e linear da reta de regressão. Para o IP6, a faixa abrangeu as concentrações de 30 µg a 240 µg, enquanto as do ácido oxálico variaram de 0,1 µg a 8 µg.
2.6.3 - Repetitividade do equipamento
A repetitividade de injeção do equipamento foi demonstrada estatisticamente pela verificação da presença de "outlyers" mediante o teste de Grubbs [32] e pelos valores dos coeficientes de variação (CVr) das áreas e dos tempos de retenção. Para isto, injetou-se 9 vezes a solução padrão correspondente a 90 µg de IP6 ou 0,8 µg de ácido oxálico. Foram calculadas, também, a média, o desvio padrão e a mediana. O teste foi repetido após 15 dias.
2.6.4 - Recuperação do analito
A eficiência do método de purificação foi determinada mediante o cálculo da porcentagem de recuperação dos ácidos fítico ou oxálico, segundo o seguinte procedimento:
A recuperação foi estimada segundo o seguinte cálculo:

em que C1 = concentração determinada na amostra adicionada; C2 = concentração determinada na amostra não adicionada; C3 = concentração adicionada.
Foram calculadas as médias, os desvios-padrão, as medianas e as presenças de "outlyers", utilizando o teste de Grubbs [32], as recuperações R(%) e os coeficientes de repetitividade (CVr) dos resultados de cada concentração.
Após o cálculo da recuperação média M (média de todas as recuperações das 3 concentrações) e do desvio-padrão relativo (CV%) das médias, verificou-se se a recuperação média diferiu ou não de 100% por meio da seguinte fórmula:

Observar que se tomou ttab para p < 0,05 e v = (n - 1), sendo n = 21 determinações [33]. No caso do tcal ser maior que o ttab, então a recuperação diferiu estatisticamente de 100%.
2.6.5 - Limites de detecção e quantificação do equipamento
Os limites do equipamento para detecção (LDE) e quantificação (LQE) foram estabelecidos em seguida [36]. Tendo-se realizado o cálculo do coeficiente angular da reta de calibração (b), obteve-se a nova curva de calibração injetando-se, em triplicata, o padrão de hexafosfato de inositol nas concentrações de 3, 6 e 15 µg ou de ácido oxálico (0,01; 0,02 e 0,04 µg), nos 20 µL injetados. Com base na equação da reta obtida para cada padrão, determinou-se a interseção com o eixo Y, ou seja, o valor do coeficiente linear da reta (Ybl). A seguir, determinaram-se os desvios-padrão das replicatas de cada concentração e construiu-se a curva de concentração x desvio-padrão e, como no caso anterior, obteve-se o coeficiente linear desta nova reta (Sbl). Os limites foram calculados como:

2.6.6 - Limites de detecção e quantificação do método
Em seguida, os limites de detecção (LDM) e de quantificação (LQM) do método (em µg) foram calculados levando-se em conta o LDE e o LQE, corrigidos pela porcentagem de recuperação do nível inferior do padrão adicionado.
3 - RESULTADOS E DISCUSSÃO
3.1 - Determinação do ácido fítico
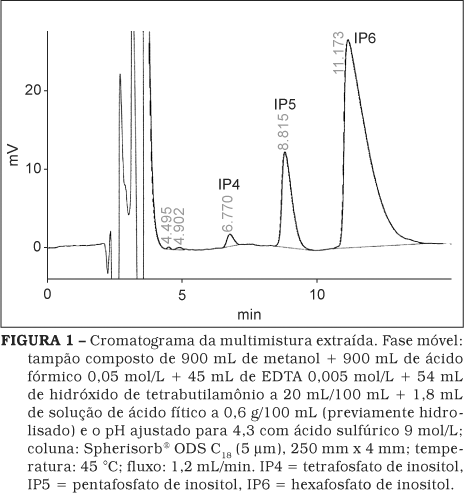
Como pode ser observado na Tabela 1, o padrão contém como impureza o IP5. Ao se extrair o padrão pôde-se observar o aparecimento do derivado IP4, bem como o aumento na área do IP5 e a diminuição da área do IP6, ficando evidente que ocorreu a hidrólise deste último e a formação dos demais. Esta hidrólise pode ser explicada pelas condições de extração da matriz que ocorre em meio aquoso ácido e sob temperaturas relativamente elevadas. Assim, nas amostras de multimistura extraída (Figura 1) e multimistura adicionada de IP6 e extraída (Tabela 1), pode-se constatar a presença destes derivados de IP6 e, inclusive, o aparecimento na última do derivado IP3. A presença deste derivado pôde ser explicada pelo fato da concentração de ácido fitico presente na amostra (natural + adicionado) ter sido suficiente para que a hidrólise do IP6 e, provavelmente dos IP5 e IP4, ter produzido IP3 em nível detectável pelo método. Assim, a quantificação do IP6 foi feita interpolando-se a área da amostra com a equação da curva do padrão não-extraído. A determinação do fitato total foi realizada levando-se em conta a soma das áreas do IP6 com as dos IP5 e IP4, corrigidas por um fator de proporcionalidade [10].
3.1.1 - Seletividade e especificidade
Não se constatou a presença de picos interferentes nos cromatogramas obtidos com o detector de índice de refração. Contudo, não foi possível obter um espectro de varredura por arranjo de diodos característico para o ácido fítico e seus derivados uma vez que estes absorvem numa região do ultravioleta abaixo do alcance do detector. Deste modo, considerando-se apenas os valores das áreas obtidas do branco, dos padrões e da amostra adicionada, a simetria dos picos e a concordância dos tempos de retenção, concluiu-se que não houve evidências da presença de substâncias interferentes junto aos picos do ácido fítico e seus derivados.
Além disto, avaliou-se também a influência da matriz na recuperação do ácido fítico comparando-se a inclinação da reta do padrão de ácido fítico extraído com a do padrão adicionado à multimistura. O valor obtido para a curva do padrão adicionado (b = 28089) está fora dos limites de confiança da curva do padrão extraído (24627 < b < 26043), ou seja, estatisticamente pode-se considerar que a matriz influencia na recuperação do ácido fítico.
3.1.2 - Linearidade
A equação da reta obtida pelo método dos mínimos quadrados é representada pela fórmula: (Y = 28761X 128449) e o coeficiente de correlação (r) foi de 0,99964. A linearidade da curva do ácido fítico foi demonstrada estatisticamente pelo valor de tcal (135,36) que foi maior que o valor de ttab (2,447), para n = 7 e p < 0,05.
3.1.3 - Repetitividade
Os valores de Grubbs máximos e mínimos calculados para as áreas (1,63 e 1,44, respectivamente) e para os tempos de retenção (1,23 e 1,70, respectivamente) estão abaixo do valor crítico de 2,22 (para n = 9 e p < 0,05), ou seja, não foram encontrados "outlyers". Os valores dos CVr das áreas e dos tempos de retenção encontrados (1,29 e 0,15, respectivamente) foram menores que o valor crítico de 2,7 (correspondente a analitos presentes em concentrações de 1 g/100 g de amostra) [36].
3.1.4 - Recuperação do ácido fítico
A recuperação do ácido fítico adicionado à amostra (IP6 11,173 min) variou de 43,6% para o nível baixo de adição, passando para 78,1% para o médio e chegando a 86,0% no alto. Se ainda forem consideradas as áreas dos picos dos derivados IP5, IP4 (Figura 1), as recuperações corresponderam a 49,2%, 91,3%, 101,0%, respectivamente. Estas últimas foram utilizadas no cálculo das concentrações das amostras conforme o nível encontrado. Como referência também foi injetado o padrão de hexafosfato de inositol (Tabela 1), podendo-se observar a presença de IP5 (9,151 min) como contaminante do padrão de IP6.
Com base nos resultados obtidos, pôde-se inferir que um ou mais componentes da multimistura interferem na recuperação do fitato, principalmente quando em concentrações mais baixas. Os prováveis interferentes seriam minerais, principalmente o cálcio e o ferro, presentes na casca de ovo e nos demais ingredientes. Fato semelhante já havia sido constatado quando dois métodos de doseamento de ácido fítico por CLAE foram comparados [6] por pareamento iônico e outro por exclusão iônica. Os valores de recuperação encontrados foram 14 a 190 vezes maiores no segundo método em relação ao primeiro. Estes resultados haviam sido obtidos em amostras de preparados de cereais infantis, que por sua composição apresentam concentrações baixas de ácido fítico e elevadas de minerais.
3.1.5 - Limites de detecção e quantificação
Os limites de detecção e quantificação do equipamento foram de 2,59 e 6,41 µg de ácido fítico nos 20 µL injetados, respectivamente. Uma vez que a recuperação do ácido fítico diferiu estatisticamente de 100%, os LDM e LQM foram corrigidos em relação aos do equipamento, resultando em 2,74 e 6,78 µg nos 20 µL injetados, respectivamente.
3.2 - Determinação do ácido oxálico
A determinação do ácido oxálico por CLAE de fase reversa [27], inicialmente, foi realizada empregando-se a fase móvel composta do tampão contendo fosfato de potássio monobásico 0,5 g/100 mL e hidróxido de tetrabutilamônio 0,005 mol/L. O pH foi ajustado para 2,0 adicionando-se ácido ortofosfórico. Como fase estacionária, foi utilizada a coluna de fase reversa C18 termoestatizada a 40 °C por 1 h. A vazão variou de 0,5 a 2,0 mL/min para um volume injetado de 20 µL. Entretanto este procedimento não apresentou resultados satisfatórios quanto à simetria do pico alargado e com a linha de base irregular (Tr = 4,366 min, área = 1113587, alt. = 53018 mAbs). Posteriormente, trocou-se a coluna por uma de fase reversa C8 mantendo-se a fase móvel, mas continuou a ocorrer o mesmo problema. Nos dois procedimentos, foi usado o detector UV/VIS ajustado para leitura no comprimento de onda de 220 nm.
Assim, resolveu-se adicionar o hidróxido de tetrametilamônio ao tampão (pareamento iônico) a fim de melhorar a simetria do pico e a linha de base. O resultado obtido foi satisfatório para o padrão puro (Tr = 4,898 min, área = 2154366, alt. = 217959 mAbs). Contudo, ao se aplicar o método à amostra da multimistura, não foi possível obter uma separação completa do pico do ácido oxálico de um pico desconhecido retido aos 5,134 min (Figura 2), embora se tivesse condições de calcular a área do pico do ácido oxálico (Tr = 4,877 min, área = 173795, alt. = 16976 mAbs). Cumpre ressaltar que ao se analisar um extrato do pó de folha de mandioca puro fonte em potencial do ácido oxálico este pico interferente não apareceu no cromatograma.
3.2.1 - Seletividade e especificidade
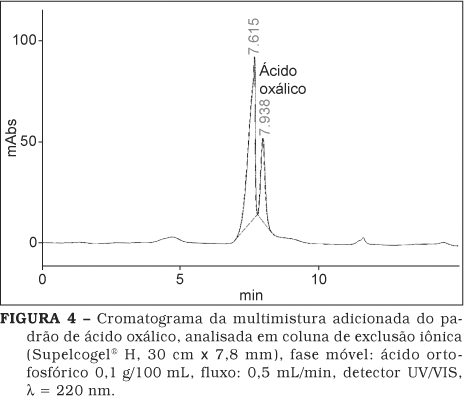
Avaliou-se o emprego da cromatografia de exclusão iônica com o DAD. Uma vez que o pico obtido do ácido oxálico, tanto no padrão como nas amostras, estava suficientemente separado dos interferentes (Figuras 3 e 4), concluiu-se que este último método analítico seria o mais adequado para a análise das amostras da multimistura.
Além disto, avaliou-se também a influência da matriz na recuperação do ácido oxálico comparando-se a inclinação da reta do padrão não-extraído com a do padrão adicionado à multimistura. O valor obtido para a curva do padrão adicionado (b = 614296) está fora dos limites de confiança da curva do padrão não-adicionado (677645 < b < 685157), ou seja, estatisticamente pode-se considerar que a matriz influencia na recuperação do ácido oxálico.
3.2.2 - Linearidade
A curva de calibração do padrão de ácido oxálico obtida segundo o método descrito é representada pela equação da reta obtida pelo método dos mínimos quadrados, em que Y = 689213x 15516 e o coeficiente de correlação (r) foi de 0,99985. Assim, a linearidade da curva do ácido oxálico foi demonstrada estatisticamente pelo valor de tcal (198,97) que foi maior que o valor de ttab (2,447) para n = 7 e p < 0,05.
3.2.3 - Repetitividade
Os valores de Grubbs máximos e mínimos encontrados para os tempos de retenção (1,62 e 1,30, respectivamente) foram menores do que o valor crítico de 2,22 (para n = 9 e p < 0,05). Os valores dos CVr das áreas e dos tempos de retenção encontrados (0,590 e 0,039, respectivamente) foram menores que o valor crítico de 2,7 (correspondente a analitos presentes em concentrações de 1 g/100 g da amostra) [36].
3.2.4 - Recuperação do método
A recuperação do ácido oxálico variou de 60,6% para o nível baixo de adição, passando para 73,4% para o nível médio e chegando a 87,6% para o nível alto. Estas foram utilizadas no cálculo das concentrações nas amostras conforme o nível encontrado,
Observou-se que a recuperação do ácido oxálico tende a ser maior nos níveis mais altos do que nos mais baixos. Provavelmente, estes resultados se devem à interferência de uma substância com tempo de retenção próximo ao do ácido oxálico e com espectro de absorção semelhante ao deste ácido, principalmente na região de 200 a 220 nm.
3.2.5 - Limites de detecção e quantificação
Os limites de detecção e quantificação do equipamento obtidos foram de 0,0019 µg e 0,0057 µg de ácido oxálico, respectivamente, nos 20 µL injetados. Uma vez que a recuperação do ácido oxálico diferiu estatisticamente de 100%, os LDM e LQM foram corrigidos em relação aos do equipamento, resultando em 0,00227 µg e 0,00681 µg, nos 20 µL injetados, respectivamente.
3.3 - Teores dos ácidos fítico e oxálico na multimistura
As análises dos constituintes objetos deste estudo, feitas nas 30 amostras de multimistura coletadas no período de outubro de 2002 a junho de 2003, resultaram nas seguintes faixas de concentrações para o ácido fítico: de 1,61 ± 0,26 a 2,25 ± 0,09 g/100 g; e para o ácido oxálico: de 0,044 ± 0,005 a 0,057 ± 0,003 g/100 g (Tabela 2). As médias globais corresponderam a 2,01 ± 0,27 g/100 g para o ácido fítico e 0,051 ± 0,005 para o ácido oxálico. Assim, pôde-se constatar que os teores de ácido fítico encontrados estão cerca de vinte vezes acima do limite máximo permitido pelo Regulamento Técnico de Identidade e Qualidade de Mistura à Base de Farelos de Cereais, que é de 0,1 g/100 g da amostra [5].
4 - CONCLUSÕES
Com relação ao ácido fítico pôde-se concluir que:
Com relação ao ácido oxálico pôde-se concluir que:
5 - REFERÊNCIAS BIBLIOGRÁFICAS
[1] ALENCAR, M. L. C. B.; SILVESTRE, M. P. C. Métodos de determinação de oxalatos em vegetais. In: ENCONTRO NACIONAL DE ANALISTAS DE ALIMENTOS, 6, Curitiba, 1990. Resumos... Curitiba: Instituto de Tecnologia do Paraná, 1990. p. 103.
[2] ASSOCIATION OF OFFICIAL ANALYTICAL CHEMISTS. Official methods of analysis. 15. ed. Arlington: AOAC, 1990. (Met. 986.11.).
[3] BAKER, C. J. L. [Dosagem de ácido oxálico]Analyst (London), v. 77, p. 340, 1952 apud WILSON, C.W.; SHAW, P.E.; KNIGHT Jr., R.J. Analysis of oxalic acid in carambola (Averrhoa carambola L.) and spinach by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1106-1108, 1982.
[4] BRANDÃO, C. T. Alternativas alimentares. Brasília: Ministério da Saúde, Divisão Nacional de Educação para a Saúde - Pastoral da Criança, 1988. 49 p.
[5] BRASIL. Agência Nacional de Vigilância Sanitária. Resolução n. 53, de 15 de junho de 2000. Dispõe sobre o Regulamento Técnico de Identidade e Qualidade de Mistura à base de Farelo de Cereais. Diário Oficial [da] República Federativa do Brasil, Brasília, DF, 19 jun. 2000. p. 1.
[6] BROOKS, S. P. J.; LAMPI, B. J. Problems associated with measuring phytate in infant cereals. J. Agric. Food Chem., v. 49, n. 2, p. 564-569, 2001.
[7] BUSHWAY, R. J.; BUREAU, J. L.; MCGANN, D. F. Determinations of organic acids in potatoes by high performance liquid chromatography. J. Food Sci., v. 49, n. 1, p. 75-77, 1984.
[8] CÂMARA, F. S.; MADRUGA, M. S. Conteúdos de ácido cianídrico, ácido fítico, tanino total e aflatoxina em uma preparação brasileira (Natal) de multimistura. Rev. Nutr., v. 14, n. 1, p. 33-36, 2001.
[9] CAMIRE, A. L.; CLYDESDALE, F. M. Analysis of phytic acid in foods by HPLC. J. Food Sci., v. 47, n. 3, p. 575-578, 1982.
[10] CUADRADO, C.; MUZQUIZ, M.; ROBREDO, L. M.; AYET, G.; BURBANO, C. Determinación de factores antinutritivos termorresistentes en leguminosas. III. Fitato. Invest. Agrar.: Prod. Protec. Veg., v. 10, n. 2, p. 157-165, 1995.
[11] DEBESSAUTET, I. Estudio de las bases cientificas para el uso de alimentos alternativos en la nutrición humana. Brasilia: INAN, 1992. 92 p.
[12] FRANCO, V.; KRINITZ, H. Determination of oxalic acid in foods. J. Assoc. Off. Agric. Chem., v. 56, n. 1, p. 164-166, 1973.
[13] FURTUNATO, D. M. N. Multimistura: sua relação químico-nutricional. 2003. 63 f. Monografia (Doutorado em Química Analítica). Instituto de Química da UFBA. Salvador. 2003.
[14] GOTTSTEIN, H. D.; ZOOK, M. N.; KUC, J. A. Detection and quantitation of oxalic acid by capillary gas chromatography. J. Chromatogr., v. 481, p. 55-61, 1989.
[15] GRAF, E.; DINTZIS, F. R. Determination of phytic acid in foods by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1094-1097, 1982.
[16] HARLAND, B. F.; OBERLEAS, D. Anion-exchange method for determination of phytate in foods: collaborative study. J. Ass. Off. Anal. Chem., v. 69, n. 4, p. 667-670, 1986.
[17] HEUBNER, W.; STADLER, H. Determination of phytin by titration. Biochem. Z., v. 64, n. 8, p. 422, 1914.
[18] HODGKINSON, A. Oxalic acid in biology and medicine. London: Academic Press, 1977. 345 p.
[19] HONOW, R.; BONGARTZ, D.; HESSE, A. An improved HPLC-enyme-reactor method for the determination of oxalic acid in complex matrices. Clin. Chim. Acta, v. 261, n. 2, p. 131-139, 1997.
[20] INMETRO Normalização e qualidade industrial - Orientações sobre validação de métodos e ensaios químicos. DOQ-CGCRE-008, outubro 2002. Disponível em: < http://www.inmetro.gov.br/kits/docqre008r01.pdf> Acesso em: 01 jul. 2003.
[21] INSTITUTO ADOLFO LUTZ. Normas analíticas do Instituto Adolfo Lutz - métodos químicos e físicos para análise de alimentos. 3. ed. São Paulo: IAL, 1985. (Met. 6.1.1.2.).
[22] KNUCKLES, B. E.; KUZMICKY, D. D.; BETSCHART, A. A. HPLC analysis of phytic acid in selected foods and biological samples. J. Food Sci., v. 49, n. 4, p. 1257-1258, 1982.
[23] LEE, K.; ABENDROTH, J. A. High performance liquid chromatographic determination of phytic acid in foods. J. Food Sci., v. 48, n. 4, p. 1344-1349, 1983.
[24] LEHRFELD, J. High-performance liquid chromatography analysis of phytic acid on a pH-stable, macroporous polymer column. Cereal Chem., v. 66, n. 6, p. 510-515, 1989.
[25] LEHRFELD, J.; MORRIS, E. R. Overestimation of phytic acid in foods by the AOAC anion-exchange method. J. Agric. Food Chem., v. 40, n. 11, p. 2208-2210, 1992.
[26] LIAN, H. Z.; MAO, L.; YE, X. L.; MIAO, J. Simultaneous determination of oxalic, fumaric, maleic and succinic acids in tartaric and malic acids for pharmaceutical use by ion-suppression reversed-phase high performance liquid chromatography. J. Pharm. Biom. Anal., v. 19, n. 3-4, p. 621-625, 1999.
[27] LIBERT, B. Rapid determination of oxalic acid by reversed-phase high-performance liquid chromatography. J. Chromatogr., v. 210, n. 3, p. 540-543, 1981.
[28] LOURES, A.; JOKL, L. Microtécnica para determinação de ácido oxálico em folhas e derivados. In: ENCONTRO NACIONAL DE ANALISTAS DE ALIMENTOS, 6, Curitiba, 1990. Resumos... Curitiba: Instituto de Tecnologia do Paraná, 1990. p. 59.
[29] MADRUGA, M. S.; CÂMARA, F. S. The chemical composition of "multimistura" as a food supplement. Food Chem., v. 68, n. 1, p. 41-44, 2000.
[30] MAYR, G. W. A novel metal-dye detection system permits picomolar-range h.p.l.c. analysis of inositol polyphosphates from non-radioactively labelled cell or tissue specimens. Biochem. J., v. 254, n. 2, p. 585-591, 1988.
[31] MENDENHALL, W.; OTT, L.; SHAEAFFER, R. L. Elementary survey sampling. Boston: Wadsworth, 1986. 320 p.
[32] NETO, B. B.; SCARMINIO, I. S.; BRUNS, R. E. Como fazer experimentos: pesquisa e desenvolvimento na ciência e na indústria. Campinas: Universidade de Campinas, 2001. 401 p.
[33] PIMENTEL-GOMES, F. Curso de estatística experimental. 13. ed. São Paulo: Nobel, 1990. 467 p.
[34] POTEZNY, N.; BAIS, R. J.; O'LOUGHLIN, P. I.; EDWARDS, J. B.; ROFE, A. M. Urinary oxalate determination by use of immobilized oxalate oxidase in a continuous-flow system. Am. J. Clin. Chem., v. 29, n. 1, p. 1620, 1983.
[35] PUCHER, G. W.; VICKERY, H. B.; WAKEMAN, A. J. J. [Dosagem de ácido oxálico]. Ind. Eng. Chem., v. 6, p. 140, 1934 apud WILSON, C. W.; SHAW, P. E.; KNIGHT Jr., R. J. Analysis of oxalic acid in carambola (Averrhoa carambola L.) and spinach by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1106-1108, 1982.
[36] QUATROCCHI, O. A.; ANDRIZZI, S. A.; LABA, R. F. Introducción a la HPLC: aplicación y práctica. Buenos Aires: Artes Gráficas Farro, 1992. p. 301-328.
[37] ROSS, A. B.; SAVAGE, G. P.; MARTIN, R. J.; VANHANEN, L. Oxalates in oca (New Zealand yam) (Oxalis tuberosa Mol.). J. Agric. Food Chem., v. 47, n. 12, p. 5019-5022, 1999.
[38] ROUNDS, M. A.; NIELSEN, S. S. Anion-exchange high performance liquid chromatography with postcolumn detection for analysis of phytic acid and other inositol phosphates. J. Chromatogr. A, v. 653, n. 1, p. 148-152, 1993.
[39] SANDBERG, A. S.; AHDERINNE, R. HPLC method for determination of inositol tri-, tetra-, penta- and hexaphosphates in foods and intestinal contents. J. Food Sci., v. 51, n. 3, p. 547-550, 1986.
[40] SANDBERG, A. S.; CARLSON, N. G.; SVANBERG, U. Effects of inositol tri-, tetra-, penta-, and hexaphosphates on in vitro estimation of iron availability. J. Food Sci., v. 54, n. 1, p. 159-161, 1989.
[41] SANT'ANA, L. F. R.; COSTA, N. M. B.; FERREIRA, G.; OLIVEIRA, M. G. A.; MONTEIRO, J. B. R. Valor nutritivo e fatores antinutricionais de multimisturas utilizadas como alternativa alimentar. Braz. J. Food Technol., v. 3, n. 45, p. 129-135, 2000.
[42] SARKAR, S. K.; MALHOTRA, S. S. Gas-liquid chromatographic method for separation of organic acids and its application of pine needle extracts. J. Chromatogr., v. 171, p. 227-232, 1 april 1979.
[43] SAVAGE, G. P.; VANHANEN, L.; MASON, S. L.; ROSS, A. B. Effects of cooking on the soluble and insoluble oxalate content of some New Zealand foods. J. Food Comp. Anal., v. 13, n. 3, p. 201-206, 2000.
[44] SILVA, B. M.; ANDRADE, P. B.; MENDES, G. C.; SEABRA, R. M.; FERREIRA, M. A. Study of organic acids composition of quince (Cydonia oblonga miller) fruit and jam. J. Agric. Food Chem., v. 50, n. 8, p. 23132317, 2002.
[45] SNEDECOR, G. W.; COCHRAN, W. G. Métodos estatísticos. Mexico CD: Continental, 1980. p. 611-653.
[46] SOUZA, M. S.; LIMA, V. L. A. G. Multimistura: aspectos químicos, microbiológicos e nutricionais. Nutr. Brasil, v. 4, n. 2, p. 93-98, 2005.
[47] VAN HEES, P. A. W.; DAHLÉN, J.; LUNDSTROM, U. S.; BORÉN, H.; ALLARD, B. Determination of low molecular weigth organic acids in soil solution by HPLC. Talanta, v. 48, n. 1, p. 173-179, 1999.
[48] WILSON, C. W.; SHAW, P. E.; KNIGHT Jr., R. J. Analysis of oxalic acid in carambola (Averrhoa carambola L.) and spinach by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1106-1108, 1982.
[49] XU, P.; PRICE, J.; AGGETT, P. J. Recent advances in methodology for analysis of phytate and inositol phosphates in foods. Prog. Food Nutr. Sci., v. 16, n. 3, p. 245-262, 1992.
[50] YANG, L.; LIU, L.; OLSEN, B. A.; NUSSBAUM, M. A. The determination of oxalic acid, oxamic acid and oxamide in a drug substance by ion-exclusion chromatography. J. Pharmaceut. Biomed. Anal., v. 22, n. 3, p. 487-493, 2000.
[51] ZAREMBSKI, P. M.; HODGKINSON, A. The determination of oxalic acid in food. Analyst (London), v. 87, n. 1038, p. 698-702, 1962.
Recebido para publicação em 5/12/2005.
Aceito para publicação em 20/10/2006 (001646).
Parte da dissertação de Mestrado em Ciência de Alimentos da Faculdade de Farmácia da Universidade Federal de Minas Gerais, do 1º autor.
- [1] ALENCAR, M. L. C. B.; SILVESTRE, M. P. C. Métodos de determinação de oxalatos em vegetais. In: ENCONTRO NACIONAL DE ANALISTAS DE ALIMENTOS, 6, Curitiba, 1990. Resumos.. Curitiba: Instituto de Tecnologia do Paraná, 1990. p. 103.
- [2] ASSOCIATION OF OFFICIAL ANALYTICAL CHEMISTS. Official methods of analysis 15. ed. Arlington: AOAC, 1990. (Met. 986.11.).
- [3] BAKER, C. J. L. [Dosagem de ácido oxálico]Analyst (London), v. 77, p. 340, 1952 apud WILSON, C.W.; SHAW, P.E.; KNIGHT Jr., R.J. Analysis of oxalic acid in carambola (Averrhoa carambola L.) and spinach by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1106-1108, 1982.
- [4] BRANDÃO, C. T. Alternativas alimentares Brasília: Ministério da Saúde, Divisão Nacional de Educação para a Saúde - Pastoral da Criança, 1988. 49 p.
- [5] BRASIL. Agência Nacional de Vigilância Sanitária. Resolução n. 53, de 15 de junho de 2000. Dispõe sobre o Regulamento Técnico de Identidade e Qualidade de Mistura à base de Farelo de Cereais. Diário Oficial [da] República Federativa do Brasil, Brasília, DF, 19 jun. 2000. p. 1.
- [6] BROOKS, S. P. J.; LAMPI, B. J. Problems associated with measuring phytate in infant cereals. J. Agric. Food Chem., v. 49, n. 2, p. 564-569, 2001.
- [7] BUSHWAY, R. J.; BUREAU, J. L.; MCGANN, D. F. Determinations of organic acids in potatoes by high performance liquid chromatography. J. Food Sci., v. 49, n. 1, p. 75-77, 1984.
- [8] CÂMARA, F. S.; MADRUGA, M. S. Conteúdos de ácido cianídrico, ácido fítico, tanino total e aflatoxina em uma preparação brasileira (Natal) de multimistura. Rev. Nutr., v. 14, n. 1, p. 33-36, 2001.
- [9] CAMIRE, A. L.; CLYDESDALE, F. M. Analysis of phytic acid in foods by HPLC. J. Food Sci., v. 47, n. 3, p. 575-578, 1982.
- [10] CUADRADO, C.; MUZQUIZ, M.; ROBREDO, L. M.; AYET, G.; BURBANO, C. Determinación de factores antinutritivos termorresistentes en leguminosas. III. Fitato. Invest. Agrar.: Prod. Protec. Veg., v. 10, n. 2, p. 157-165, 1995.
- [11] DEBESSAUTET, I. Estudio de las bases cientificas para el uso de alimentos alternativos en la nutrición humana Brasilia: INAN, 1992. 92 p.
- [12] FRANCO, V.; KRINITZ, H. Determination of oxalic acid in foods. J. Assoc. Off. Agric. Chem., v. 56, n. 1, p. 164-166, 1973.
- [13] FURTUNATO, D. M. N. Multimistura: sua relação químico-nutricional 2003. 63 f. Monografia (Doutorado em Química Analítica). Instituto de Química da UFBA. Salvador. 2003.
- [14] GOTTSTEIN, H. D.; ZOOK, M. N.; KUC, J. A. Detection and quantitation of oxalic acid by capillary gas chromatography. J. Chromatogr., v. 481, p. 55-61, 1989.
- [15] GRAF, E.; DINTZIS, F. R. Determination of phytic acid in foods by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1094-1097, 1982.
- [16] HARLAND, B. F.; OBERLEAS, D. Anion-exchange method for determination of phytate in foods: collaborative study. J. Ass. Off. Anal. Chem., v. 69, n. 4, p. 667-670, 1986.
- [17] HEUBNER, W.; STADLER, H. Determination of phytin by titration. Biochem. Z., v. 64, n. 8, p. 422, 1914.
- [18] HODGKINSON, A. Oxalic acid in biology and medicine London: Academic Press, 1977. 345 p.
- [19] HONOW, R.; BONGARTZ, D.; HESSE, A. An improved HPLC-enyme-reactor method for the determination of oxalic acid in complex matrices. Clin. Chim. Acta, v. 261, n. 2, p. 131-139, 1997.
- [20] INMETRO Normalização e qualidade industrial - Orientações sobre validação de métodos e ensaios químicos. DOQ-CGCRE-008, outubro 2002. Disponível em: < http://www.inmetro.gov.br/kits/docqre008r01.pdf> Acesso em: 01 jul. 2003.
- [21] INSTITUTO ADOLFO LUTZ. Normas analíticas do Instituto Adolfo Lutz - métodos químicos e físicos para análise de alimentos. 3. ed. São Paulo: IAL, 1985. (Met. 6.1.1.2.).
- [22] KNUCKLES, B. E.; KUZMICKY, D. D.; BETSCHART, A. A. HPLC analysis of phytic acid in selected foods and biological samples. J. Food Sci., v. 49, n. 4, p. 1257-1258, 1982.
- [23] LEE, K.; ABENDROTH, J. A. High performance liquid chromatographic determination of phytic acid in foods. J. Food Sci., v. 48, n. 4, p. 1344-1349, 1983.
- [24] LEHRFELD, J. High-performance liquid chromatography analysis of phytic acid on a pH-stable, macroporous polymer column. Cereal Chem., v. 66, n. 6, p. 510-515, 1989.
- [25] LEHRFELD, J.; MORRIS, E. R. Overestimation of phytic acid in foods by the AOAC anion-exchange method. J. Agric. Food Chem., v. 40, n. 11, p. 2208-2210, 1992.
- [26] LIAN, H. Z.; MAO, L.; YE, X. L.; MIAO, J. Simultaneous determination of oxalic, fumaric, maleic and succinic acids in tartaric and malic acids for pharmaceutical use by ion-suppression reversed-phase high performance liquid chromatography. J. Pharm. Biom. Anal., v. 19, n. 3-4, p. 621-625, 1999.
- [27] LIBERT, B. Rapid determination of oxalic acid by reversed-phase high-performance liquid chromatography. J. Chromatogr., v. 210, n. 3, p. 540-543, 1981.
- [28] LOURES, A.; JOKL, L. Microtécnica para determinação de ácido oxálico em folhas e derivados. In: ENCONTRO NACIONAL DE ANALISTAS DE ALIMENTOS, 6, Curitiba, 1990. Resumos.. Curitiba: Instituto de Tecnologia do Paraná, 1990. p. 59.
- [29] MADRUGA, M. S.; CÂMARA, F. S. The chemical composition of "multimistura" as a food supplement. Food Chem., v. 68, n. 1, p. 41-44, 2000.
- [30] MAYR, G. W. A novel metal-dye detection system permits picomolar-range h.p.l.c. analysis of inositol polyphosphates from non-radioactively labelled cell or tissue specimens. Biochem. J., v. 254, n. 2, p. 585-591, 1988.
- [31] MENDENHALL, W.; OTT, L.; SHAEAFFER, R. L. Elementary survey sampling Boston: Wadsworth, 1986. 320 p.
- [32] NETO, B. B.; SCARMINIO, I. S.; BRUNS, R. E. Como fazer experimentos: pesquisa e desenvolvimento na ciência e na indústria. Campinas: Universidade de Campinas, 2001. 401 p.
- [33] PIMENTEL-GOMES, F. Curso de estatística experimental 13. ed. São Paulo: Nobel, 1990. 467 p.
- [34] POTEZNY, N.; BAIS, R. J.; O'LOUGHLIN, P. I.; EDWARDS, J. B.; ROFE, A. M. Urinary oxalate determination by use of immobilized oxalate oxidase in a continuous-flow system. Am. J. Clin. Chem., v. 29, n. 1, p. 1620, 1983.
- [35] PUCHER, G. W.; VICKERY, H. B.; WAKEMAN, A. J. J. [Dosagem de ácido oxálico]. Ind. Eng. Chem., v. 6, p. 140, 1934
- apud WILSON, C. W.; SHAW, P. E.; KNIGHT Jr., R. J. Analysis of oxalic acid in carambola (Averrhoa carambola L.) and spinach by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1106-1108, 1982.
- [36] QUATROCCHI, O. A.; ANDRIZZI, S. A.; LABA, R. F. Introducción a la HPLC: aplicación y práctica Buenos Aires: Artes Gráficas Farro, 1992. p. 301-328.
- [37] ROSS, A. B.; SAVAGE, G. P.; MARTIN, R. J.; VANHANEN, L. Oxalates in oca (New Zealand yam) (Oxalis tuberosa Mol.). J. Agric. Food Chem., v. 47, n. 12, p. 5019-5022, 1999.
- [38] ROUNDS, M. A.; NIELSEN, S. S. Anion-exchange high performance liquid chromatography with postcolumn detection for analysis of phytic acid and other inositol phosphates. J. Chromatogr. A, v. 653, n. 1, p. 148-152, 1993.
- [39] SANDBERG, A. S.; AHDERINNE, R. HPLC method for determination of inositol tri-, tetra-, penta- and hexaphosphates in foods and intestinal contents. J. Food Sci., v. 51, n. 3, p. 547-550, 1986.
- [40] SANDBERG, A. S.; CARLSON, N. G.; SVANBERG, U. Effects of inositol tri-, tetra-, penta-, and hexaphosphates on in vitro estimation of iron availability. J. Food Sci., v. 54, n. 1, p. 159-161, 1989.
- [41] SANT'ANA, L. F. R.; COSTA, N. M. B.; FERREIRA, G.; OLIVEIRA, M. G. A.; MONTEIRO, J. B. R. Valor nutritivo e fatores antinutricionais de multimisturas utilizadas como alternativa alimentar. Braz. J. Food Technol., v. 3, n. 45, p. 129-135, 2000.
- [42] SARKAR, S. K.; MALHOTRA, S. S. Gas-liquid chromatographic method for separation of organic acids and its application of pine needle extracts. J. Chromatogr., v. 171, p. 227-232, 1 april 1979.
- [43] SAVAGE, G. P.; VANHANEN, L.; MASON, S. L.; ROSS, A. B. Effects of cooking on the soluble and insoluble oxalate content of some New Zealand foods. J. Food Comp. Anal., v. 13, n. 3, p. 201-206, 2000.
- [44] SILVA, B. M.; ANDRADE, P. B.; MENDES, G. C.; SEABRA, R. M.; FERREIRA, M. A. Study of organic acids composition of quince (Cydonia oblonga miller) fruit and jam. J. Agric. Food Chem., v. 50, n. 8, p. 23132317, 2002.
- [45] SNEDECOR, G. W.; COCHRAN, W. G. Métodos estatísticos Mexico CD: Continental, 1980. p. 611-653.
- [46] SOUZA, M. S.; LIMA, V. L. A. G. Multimistura: aspectos químicos, microbiológicos e nutricionais. Nutr. Brasil, v. 4, n. 2, p. 93-98, 2005.
- [47] VAN HEES, P. A. W.; DAHLÉN, J.; LUNDSTROM, U. S.; BORÉN, H.; ALLARD, B. Determination of low molecular weigth organic acids in soil solution by HPLC. Talanta, v. 48, n. 1, p. 173-179, 1999.
- [48] WILSON, C. W.; SHAW, P. E.; KNIGHT Jr., R. J. Analysis of oxalic acid in carambola (Averrhoa carambola L.) and spinach by high-performance liquid chromatography. J. Agric. Food Chem., v. 30, n. 6, p. 1106-1108, 1982.
- [49] XU, P.; PRICE, J.; AGGETT, P. J. Recent advances in methodology for analysis of phytate and inositol phosphates in foods. Prog. Food Nutr. Sci., v. 16, n. 3, p. 245-262, 1992.
- [50] YANG, L.; LIU, L.; OLSEN, B. A.; NUSSBAUM, M. A. The determination of oxalic acid, oxamic acid and oxamide in a drug substance by ion-exclusion chromatography. J. Pharmaceut. Biomed. Anal., v. 22, n. 3, p. 487-493, 2000.
- [51] ZAREMBSKI, P. M.; HODGKINSON, A. The determination of oxalic acid in food. Analyst (London), v. 87, n. 1038, p. 698-702, 1962.
Datas de Publicação
-
Publicação nesta coleção
16 Abr 2007 -
Data do Fascículo
Dez 2006
Histórico
-
Aceito
20 Out 2006 -
Recebido
05 Dez 2005