Abstracts
INTRODUCTION: Mesangial cells (MC) may be involved in the glomerular alterations induced by ischemia/reperfusion injury. OBJECTIVE: To evaluate the response of immortalized MC (IMC) to 30 minutes of hypoxia followed by reoxygenation periods of 30 minutes (H/R30) or 24 hours (H/R24). METHODS: The intracellular calcium concentration ([Ca+2]i) was measured before (baseline) and after adding angiotensin II (AII, 10-5 M) in the presence and absence of glybenclamide (K ATP channel blocker). We estimated the level of intracellular ATP, nitric oxide (NO) and PGE2. RESULTS: ATP concentration decreased after hypoxia and increased after reoxygenation. Hypoxia and H/R induced increases in basal [Ca+2]i. AII induced increases in [Ca+2]i in normoxia (97 ± 9%), hypoxia (72 ± 10%) or HR30 (85 ± 17%) groups, but there was a decrease in the response to AII in group H/R24 since the elevation in [Ca+2]i was significantly lower than in control (61 ± 10%, p < 0.05). Glybenclamide did not modify this response. It was observed a significant increase in NO generation after 24 hours of reoxygenation, but no difference in PGE2 production was observed. Data suggest that H/R injury is characterized by increased basal [Ca+2]i and by an impairment in the response of cells to AII. Results suggest that the relative insensibility to AII may be at least in part mediated by NO but not by prostaglandins or vasodilator K ATP channels. CONCLUSION: H/R caused dysfunction in IMC characterized by increases in basal [Ca+2]i during hypoxia and reduction in the functional response to AII during reoxygenation.
acute kidney injury; angiotensin II; cell culture techniques; cellular hypoxia
INTRODUÇÃO: Células mesangiais (CM) podem estar envolvidas na lesão glomerular induzida por hipoxia/reperfusão (H/R). OBJETIVO: Avaliar a resposta de CM imortalizadas (CMI) à hipoxia por 30 minutos seguida de reoxigenação por 30 minutos (H/R30) ou 24 horas (H/R24). MÉTODOS: Concentração de cálcio intracelular ([Ca+2]i) foi avaliada antes (basal) e após a adição de angiotensina II (AII, 10-5 M), na presença e na ausência de glibenclamida (bloqueador de canais K ATP). Foram estimados o nível de ATP intracelular, de óxido nítrico (NO) e de PGE2. RESULTADOS: Nível de ATP diminuiu após hipóxia e aumentou após a reoxigenação. H/R induziu aumento na [Ca+2]i basal. A AII elevou a [Ca+2]i nas condições de normoxia (97 ± 9%), hipoxia (72 ± 10%) ou HR30 (85 ± 17%), porém no grupo H/R24, houve diminuição significativa na resposta à AII, uma vez que a elevação da [Ca+2]i foi mais baixa do que no controle (61 ± 10%, p < 0,05). Glibenclamida não alterou esta resposta. Houve um aumento significativo na geração de NO após 24 horas de reoxigenação, mas não foi observada nenhuma diferença na produção de PGE2. Os dados indicam que a injuria celular causada pela hipoxia/reoxigenação é caracterizada pelo aumento na [Ca+2]i basal e por uma diminuição na reatividade celular à AII. Resultados sugerem que a insensibilidade ao agonista constritor pode ser pelo menos em parte, mediada pelo NO, mas não pelas prostaglandinas ou por canais K ATP. CONCLUSÃO: H/R resultou em disfunção das CMI, caracterizada pelo aumento na [Ca+2]i basal durante a hipóxia e redução da resposta funcional a AII durante a reoxigenação.
angiotensina II; hipóxia celular; lesão renal aguda; técnicas de culturas de células
ORIGINAL ARTICLE ARTIGO ORIGINAL
The effect of hypoxia and reoxygenation in the response of mesangial cells to angiotensin II in vitro
Clara Versolato Razvickas; Fernanda Teixeira Borges; Andréia Silva de Oliveira; Nestor Schor; Mirian Aparecida Boim
Federal University of São Paulo (UNIFESP)
Correspondência para Correspondência para: Mirian Aparecida Boim Federal University of São Paulo (UNIFESP) Rua Pedro de Toledo, nº 781, 13º andar, Vila Clementino São Paulo, SP. CEP: 04039-032 E-mail: maboim@unifesp.br
ABSTRACT
INTRODUCTION: Mesangial cells (MC) may be involved in the glomerular alterations induced by ischemia/reperfusion injury.
OBJECTIVE: To evaluate the response of immortalized MC (IMC) to 30 minutes of hypoxia followed by reoxygenation periods of 30 minutes (H/R30) or 24 hours (H/R24).
METHODS: The intracellular calcium concentration ([Ca2+]i) was measured before (baseline) and after adding angiotensin II (AII, 10-5 M) in the presence and absence of glybenclamide (KATP channel blocker). We estimated the level of intracellular ATP, nitric oxide (NO) and PGE2.
RESULTS: ATP concentration decreased after hypoxia and increased after reoxygenation. Hypoxia and H/R induced increases in basal [Ca2+]i. AII induced increases in [Ca2+]i in normoxia (97 ± 9%), hypoxia (72 ± 10%) or HR30 (85 ± 17%) groups, but there was a decrease in the response to AII in group H/R24 since the elevation in [Ca2+]i was significantly lower than in control (61 ± 10%, p < 0.05). Glybenclamide did not modify this response. It was observed a significant increase in NO generation after 24 hours of reoxygenation, but no difference in PGE2 production was observed. Data suggest that H/R injury is characterized by increased basal [Ca2+]i and by an impairment in the response of cells to AII. Results suggest that the relative insensibility to AII may be at least in part mediated by NO but not by prostaglandins or vasodilator KATP channels.
CONCLUSION: H/R caused dysfunction in IMC characterized by increases in basal [Ca2+]i during hypoxia and reduction in the functional response to AII during reoxygenation.
Keywords: acute kidney injury, angiotensin II, cell culture techniques, cellular hypoxia.
INTRODUCTION
Renal ischemia remains the main cause of acute kidney injury (AKI) in adult population, presenting high rate of morbidity and mortality, as shown by a multicentre and prospective study in which approximately 60% of patients with ischemic AKI died during the hospital admission.1
Ischemic/reperfusion (I/R) injury causes substantial damage to the renal tubules, but also affects the renal microvasculature. The glomerular dysfunction observed during I/R is caused by many factors including functional response of microvasculature to hypoxia and a disequilibrium between vasoconstrictor and vasodilator substances. There is an increase in intrinsic vascular tone that reduces glomerular filtration, but also an increased response to vasoconstrictor agents, misleading the autoregulatory capacity and leaving the kidney to a vulnerable recurrent ischemic injury.2
The mechanisms of tubular damage during I/R injury are better known, however the mechanisms of glomerular injury are less understood. Mesangial cells modulate the glomerular filtration rate due to their contraction/relaxation properties in response to vasoactive substances3 directly affecting the glomerular surface area. Relaxation is a common manifestation of ischemia in smooth muscle-like cells, resulting in vasodilatation mainly dependent of K+ channels activation.4
Although the acute changes in the local concentration of oxygen have profound effects on arterial contractility, the mechanisms involved are complex and not fully characterized. However, both the releasing of endothelial factors5 and alterations in the concentration of intracellular free calcium ([Ca2+]i) seem to bear a central role in hypoxia-inducing arterial contractility.6,7 The rise of intracellular Ca+2 concentration ([Ca2+]i) has been implicated in the pathophysiology of hypoxic injury in different cell types including epithelial, endothelial and vascular smooth muscle.8,9 However the role of mesangial cells in hypoxic injury was less studied. Thus, the aim of the present study was to evaluate the functional response of mouse immortalized mesangial cells (IMC) to hypoxia/reoxygenation injury concerning to intracellular calcium levels and vasodilators factors production as nitric oxide (NO), prostaglandins (PGE2) and activation of K channels.
METHODS
Mouse immortalized mesangial cells obtained from American Type of culture Collection (ATCC), were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% bovine fetal serum, NaHCO3 2g/L, HEPES 2.6 g/L, 10.000UI/L penicillin, and 100 mg/L neomycin. Cells were maintained in the incubator at 37ºC in air mixture of 95% O2 and 5% CO2.
Four groups of cells were studied: control cells (Cont) were kept under normal oxygenation; acute hypoxia where cells were kept in gaseous mixture containing 95% N2 and 5% CO2 during 30 minutes (H30); acute hypoxia/reperfusion where cells were kept in hypoxic conditions during 30 minutes, followed by 30 minutes (H/R30) or 24 hours (H/R24) of oxygenation conditions (reoxygenation periods).
Hypoxia was induced by adding two 30 x 7 mm needles to transfix the cork and through one of these needles was connected to a system to infuse the gaseous mixture. During 30 min, 3 to 4 L per minute of this gaseous mixture was infused with the purpose to reduce the partial pressure of oxygen in the culture media.
Cell viability was assessed through the method of exclusion by utilizing two fluorescent dyes acridine orange (100 µg/mL) and ethidium bromide (100 µg/mL). The cells were examined by fluorescence light microscopy. Approximately 100 to 150 cells were counted and considered viable when they were stained green and nonviable when they were stained orange. Results were expressed as percentage (%) of viable cells.
LDH release was used to evaluate the cellular damage. The plasma membrane integrity was assessed through the leakage of the cytosolic enzyme, lactate dehydrogenase (LDH) into the culture medium. The enzyme was determined spectrophotometric in the incubation medium by measuring the rate of NADH consumption at 340 nm.
Cytosolic calcium concentration ([Ca2+]i) measurements was performed by spectrofluorometry using FURA 2-AM method. Mesangial cells (4 x 105 cells/mL) were resuspended in Thyrode solution (in mM: NaCl 137.0, KCl 2.7, CaCl2. 2H2O 1.4, MgCl2. 6 H2O 0.5, NaH2PO4.H2O 0.4, H2CO3 16.0, Glucose 5.5) supplemented with bovine serum albumin (BSA, 0.2% w/v) and incubated with fura-2 acetoxymethylester (AM; 5 µM, 23ºC) for 1h under continuous stirring. [Ca2+]i measurements were performed in a spectrofluorometer (Photon Technology International, Ontario, Canada). Maximal and minimal 340/380 rate were achieved by administration of the cell permeant digitonin (50 µM) and EGTA (4 mM) + NaOH (0.04 N) added at the end of the experiments. [Ca2+]i was calculated according to Grynkiewicz et al.10 The functional response of cells was evaluated by the change in [Ca2+]i in response to 10-5 M of angiotensin II compared to basal levels. This concentration of angiotensin II was previously determined as a concentration where [Ca2+]i reached a plateau.11
Intracellular Adenosine triphosphate (ATP) concentration was analyzed by bioluminescence through the luciferin-luciferase reaction.12 ATP concentrations in the cell lysate were quantified using an ATP-determination kit according to the manufacturer's instruction. The 96-well plates were then read (LuminuncTM Plate Nunc). A standard curve was generated with solutions of known ATP concentrations. Protein concentration of samples was determined by the method of Lowry.13 ATP levels were calculated as nanomolar ATP per milligram protein.
NO production was determined in the culture medium by the chemiluminescence method14 using the Nitric Oxide Analyzer (NOATM, Model 280; Sievers Instruments, Boulder, Co, USA). NO concentration was normalized by total cellular protein content, which was measured by the method of Lowry.
Prostaglandin (PGE2) levels were assessed by Elisa, using a commercial kit (Prostaglandin E2 enzyme immunoassay system, BIOTRAK, Amersham Pharmacia, England).15 The assay is based on competition between the PGE2 present in the sample and the fixed amount of anti-PGE2 marked with peroxidase according to the manufacture instruction.
Statistical analysis. Values were presented as mean ± SEM. ANOVA followed by Tuckey test were performed to compare differences among groups. Pared "t" test and Friedman ANOVA on Ranks were used to compare results obtained in the same group. Statistical significance was considered for p < 0.05.
RESULTS
The hypoxic condition was characterized by a significant reduction in pO2 (36 ± 3 mmHg) compared to control normoxic condition (139 ± 2 mmHg), (Figure 1A). Also the reduction in the concentration of intracellular ATP confirmed the metabolic response to the hypoxia (Figure 1B).
There was no significant difference in the cellular viability analyzed by acridine orange method among the groups control, H30, H/R30, but there was a significant decrease of the cellular viability in the group H/R24. These results were corroborated by cellular membrane permeation analyzed by intracellular enzyme LDH release. There was a significant increase in LDH in the culture medium in H/R30 (20 ± 1%) and H/R24 (41 ± 3%) in comparison to control (15 ± 1%) group (Table 1).
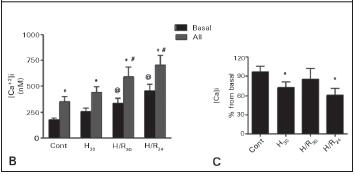
A significant increase in the basal levels of [Ca2+]i was observed in hypoxic group (H30: 365 ± 40 vs. cont: 186 ± 24 nM) and also after the reoxygention periods. (H/R30: 360 ± 73 nM and H/R24: 355 ± 48 nM), as shown in Figure 2A. Addition of AII induced an elevation in ]Cai in all groups (Figure 2B), however, the response was significantly lower in the H30 and H/R24 groups as shown in Figure 2C.

Glybenclamide induced a slight increase in [Ca2+]i which was similar among groups (Figure 3A). Glybenclamide did not change the profile of response to AII (Figure 3B), however, the intensity of increase in ]Cai induced by AII in the presence of glybenclamide was higher than that observed in the absence of glybenclamide (Figure 3C).

The NO production was analyzed in the hypoxia and hypoxia followed by reoxygenation groups. H30 and H/R30 groups did not present any difference in NO generation when compared to the control group, however, there was a significant increase in NO production in the H/R24 group compared to the normoxic conditions. In contrast, the production of PGE2 was not different among groups (Table 2).
DISCUSSION
The supply of oxygen to a tissue is largely dependent upon vascularization and the oxygenation of blood at that site, while oxygen demand is largely determined by the rate of cell respiration. Oxygen depletion can either be absolute (anoxia) or partial (hypoxia), but must be at a level below that required for complete oxidation of cytochrome C.16,17 In the present study, there was a significant decrease in pO2 and in the intracellular ATP levels indicating that the hypoxic environment was established in this in vitro model and the production of ATP was weakened. After reoxygenation the ATP levels significantly increased, although it remained bellow of the control oxygenation conditions. Also, the cellular injury and death were mainly observed after prolonged period of reoxygenation, as observed by others.18 These results point to a presence of respiratory and metabolic alterations compatible with hypoxia/reperfusion injury observed in vivo.
Mesangial cells have the contractile capacity in response to a variety of hormones, which is mediated by a rise in intracellular calcium concentration. Increases in [Ca2+]i is caused either, by Ca2+ releasing from intracellular storage sites and entry across the cell plasma membrane,19 promoting contraction. We observed a significant increase in basal [Ca2+]i levels in cells submitted to hypoxia and also to hypoxia followed by reoxygenation. This increase in [Ca2+]i may induce contraction in the mesangial cell resulting in a decrease in the glomerular filtration area, what could mediate the reduction in glomerular filtration rate during hypoxic conditions.20 The rise in [Ca2+]i could be consequent from the plasma membrane damage (increase in LDH). However, it is also important to consider that ATP depletion induced by hypoxia results in impairment of ion pumps, such as Ca-ATPase, and others transporters responsible to maintain [Ca2+]i, resulting in calcium accumulation in the cytoplasm. In addition, the increase in [Ca2+]i induced by hypoxia has also been attributed to stimulation of protein kinase C (PKC)9 probably by a reactive oxygen species (ROS)-dependent mechanism.21 Besides the contraction, the elevation in [Ca2+]i can also determine a disruption in cell homeostasis, since calcium is an important second messenger in many physiological cell responses and thus disturbances of calcium homeostasis may overwhelm the cell's capacity to maintain its physiological function.22
In contrast with increased levels of basal [Ca2+]i, the response of cells to AII was impaired after hypoxia and also after more prolonged period of reoxygenation (24h). This behavior was better observed when the [Ca2+]i was calculated as percentage of increase after adding AII. This result suggests that ischemia/reperfusion injury can modify the functional capacity of mesangial cells to react to a vasoconstrictor agent. Intracellular calcium ion is mainly regulated by a mechanism of calcium influx/efflux and Ca uptake/release by sarcoplasmic reticulum. Ca2+ influx across the plasma membrane constitutes a major component of mesangial responses to vasoconstrictors such as AII3, and thus, the reduced response to AII during H/R injury may be mainly caused by an impairment of cells to control calcium influx. In fact, Sahai et al.9 demonstrated that the calcium channel blocker verapamil prevented the hypoxia-induced stimulation of [Ca2+]i in cultured mesangial cells.
Additionally, in our experimental conditions ATP concentration decreased under hypoxia and slightly improved after reoxygenation. Recovery of cellular ATP during reperfusion depends on multiple factors that are primarily influenced by the duration of ischemia. Specific factors include the availability of ADP, AMP, nucleotide bases, and mitochondrial function. AMP provides the most rapid and major source for ATP resynthesis following in vivo ischemia.23
It is well established that during ischemia, the cellular ATP concentration significantly decreases,24 as observed in the present study. Also, it is well known that diminishing ATP/ADP ratio opens ATP sensitive potassium channel (KATP),25 resulting in cell membrane hyperpolarization and consequently closes the voltage-dependent calcium channels resulting in a decrease in [Ca2+]i. This mechanism is primarily involved in the vascular smooth muscle cell relaxation and can also decrease the sensitivity of cells to vasoconstrictors agents. Glybenclamide is a specific KATP channel blocker and addition of glybenclamide to MCs potentiated the effects of AII, although it did not change the profile of response to AII. This result suggest that KATP channels may have a role in the maintenance of the membrane resting potential in mesangial cell, but they are not involved in the impaired response of cells to AII under hyoxia/reoxygenation injury.
In order to further investigate the possible mechanism of the low reactivity of MCs to AII, we evaluated the production of two important vasodilator agents including nitric oxide and prostaglandin E2. We found no change in the PGE2 production among groups, however, there was a significant increase in the production of NO in group H/R24, suggesting that NO may be a potential mediator of the impaired response of MCs to AII during prolonged period of reoxygenation. In contrast, it has been well described that NO generation is reduced during hypoxia, causing a dysfunction of the vascular smooth muscle tone. On the other hand, the effect of hypoxia in mesangial cells in culture is poorly understood, but one possibility is that the increased NO generation under hypoxia conditions may be attributed to activation of the hypoxia-induced factor (HIF) that in turn can increase the expression of NOS and NO generation,26 however, this hypothesis must be proven in future experiments.
In conclusion, the present hypoxia/reoxygenation model caused dysfunction in IMC characterized by increases in basal [Ca2+]i after hypoxia and reoxygenation, but the response to AII was weakened after prolonged period of reoxygenation. The potential mediator of the hyporeactivity of these cells to a vasoconstrictor agent includes NO, but not KATP channels or PGE2. This functional impairment of mesangial cells may be involved in the glomerular dysfunction during ischemia and reperfusion injury.
Data de submissão: 18/06/2013.
Data de aprovação: 03/09/2013.
Capes, CNPq, FOR, FAPESP.
- 1. Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 2004;114:5-14. PMID: 15232604
- 2. Conger J. Hemodynamic factors in acute renal failure. Adv Ren Replace Ther 1997;4:25-37. PMID: 9113238
- 3. Stockand JD, Sansom SC. Glomerular mesangial cells: electrophysiology and regulation of contraction. Physiol Rev 1998;78:723-44. PMID: 9674692
- 4. Liu Y, Gutterman DD. Oxidative stress and potassium channel function. Clin Exp Pharmacol Physiol 2002;29:305-11. DOI: http://dx.doi.org/10.1046/j.1440-1681.2002.03649.x
- 5. Ward JP, Robertson TP. The role of the endothelium in hypoxic pulmonary vasoconstriction. Exp Physiol 1995;80:793-801. PMID: 8546868
- 6. Morio Y, McMurtry IF. Ca(2+) release from ryanodine-sensitive store contributes to mechanism of hypoxic vasoconstriction in rat lungs. J Appl Physiol 2002;92:527-34. PMID: 11796660
- 7. Shimizu S, Bowman PS, Thorne G 3rd, Paul RJ. Effects of hypoxia on isometric force, intracellular Ca(2+), pH, and energetics in porcine coronary artery. Circ Res 2000;86:862-70. DOI: http://dx.doi.org/10.1161/01.RES.86.8.862
- 8. Cornfield DN, Stevens T, McMurtry IF, Abman SH, Rodman DM. Acute hypoxia increases cytosolic calcium in fetal pulmonary artery smooth muscle cells. Am J Physiol 1993;265:L53-6.
- 9. Sahai A, Mei C, Pattison TA, Tannen RL. Chronic hypoxia induces proliferation of cultured mesangial cells: role of calcium and protein kinase C. Am J Physiol 1997;273:F954-60. PMID: 9435685
- 10. Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985;260:3440-50. PMID: 3838314
- 11. Hadad SJ, Ferreira AT, Oshiro ME, Neri R, Schor N. Alteration of cytosolic calcium induced by angiotensin II and norepinephrine in mesangial cells from diabetic rats. Kidney Int 1997;51:87-93. PMID: 8995721 DOI: http://dx.doi.org/10.1038/ki.1997.11
- 12. Ford SR, Leach FR. Bioluminescent assay of the guanylates. Methods Mol Biol 1998;102:55-68. PMID: 9680609
- 13. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265-75. PMID: 14907713
- 14. Hampl V, Walters CL, Archer SL. Determination of nitric oxide by the chemiluminescence reaction with ozone. In: Feelisch M, Stanler JS, eds. Methods in Nitric Oxide Research. Chichester: John Wiley & Sons; 1996. p.310-18.
- 15. Roszinski S, Jelkmann W. Effect of PO2 on prostaglandin E2 production in renal cell cultures. Respir Physiol 1987;70:131-41. PMID: 2445011
- 16. Hauser CJ, Locke RR, Kao HW, Patterson J, Zipser RD. Visceral surface oxygen tension in experimental colitis in the rabbit. J Lab Clin Med 1988;112:68-71.
- 17. Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 2003;14:2199-210. DOI: http://dx.doi.org/10.1097/01.ASN.0000079785.13922.F6
- 18. Viñas JL, Sola A, Hotter G. Mitochondrial NOS upregulation during renal I/R causes apoptosis in a peroxynitrite-dependent manner. Kidney Int 2006;69:1403-9.
- 19. Bonventre JV. Calcium and calcium-related signalling pathways in glomerular mesangial cells. Clin Exp Pharmacol Physiol 1996;23:65-70. PMID: 8713498 DOI: http://dx.doi.org/10.1111/j.1440-1681.1996.tb03064.x
- 20. Mené P, Simonson MS, Dunn MJ. Physiology of the mesangial cell. Physiol Rev 1989;69:1347-424. PMID: 2678170
- 21. Yadav VR, Song T, Joseph L, Mei L, Zheng YM, Wang YX. Important role of PLC-γ1 in hypoxic increase in intracellular calcium in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2013;304:L143-51.
- 22. Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Calcium in cell injury and death. Annu Rev Pathol 2006;1:405-34. DOI: http://dx.doi.org/10.1146/annurev.pathol.1.110304.100218
- 23. Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol 2011;22:14-20. DOI: http://dx.doi.org/10.1681/ASN.2009121217
- 24. Zager RA, Jurkowitz MS, Merola AJ. Responses of the normal rat kidney to sequential ischemic events. Am J Physiol 1985;249:F148-59. PMID: 4014471
- 25. Craig TJ, Ashcroft FM, Proks P. How ATP inhibits the open K(ATP) channel. J Gen Physiol 2008;132:131-44. DOI: http://dx.doi.org/10.1085/jgp.200709874
- 26. Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol 1998;274:L212-9. PMID: 9486205
Correspondência para:
Publication Dates
-
Publication in this collection
19 Dec 2013 -
Date of issue
Dec 2013
History
-
Received
18 June 2013 -
Accepted
03 Sept 2013