Resumos
As síndromes demenciais são caracterizadas pela presença de déficit progressivo na função cognitiva, com maior ênfase na perda de memória, e interferência nas atividades sociais e ocupacionais. O diagnóstico diferencial deve, primeiramente, identificar os quadros potencialmente reversíveis, de etiologias diversas, tais como alterações metabólicas, intoxicações, infecções, deficiências nutricionais etc. Nas demências degenerativas primárias e nas formas seqüelares, o diagnóstico etiológico carrega implicações terapêuticas e prognósticas. Sabe-se que o diagnóstico definitivo da maioria das síndromes demenciais depende do exame neuropatológico. Entretanto, uma avaliação clínica cuidadosa incluindo anamnese detalhada, exames físico e neurológico, associado a determinações bioquímicas e de neuroimagem, podem possibilitar maior acurácia no diagnóstico diferencial. Inovações tecnológicas servindo-se de métodos de neuroimagem estrutural e funcional, bem como de técnicas de biologia e genética molecular, têm apresentado perspectivas para o diagnóstico precoce das demências, particularmente da doença de Alzheimer. As diversas etiologias implicadas no desenvolvimento de síndromes demenciais, bem como as respectivas condutas diagnósticas, serão revistas neste artigo.
Demência; diagnóstico diferencial; doença de Alzheimer; demência vascular; demência não-Alzheimer
Dementia is a syndrome characterized by progressive impairment of cognitive functions, particularly in memory, which affects social and occupational activities. The differential diagnosis must, firstly, identify potentially treatable causes of cognitive impairment, addressing the different etiologies of reversible dementia such as metabolic alterations, intoxications, CNS infections, and nutritional deficiencies. The correct and early diagnosis of primary degenerative dementia carries therapeutic and prognostic implications, which may attenuate the inevitable cognitive and behavioral deficits. Definitive diagnoses of most primary dementia syndromes rely on post-mortem neuropathological examination. However, a thorough clinical evaluation, including a detailed clinical history, physical and neurological examination, combined with biochemical determinations and neuroimaging, provide a more accurate differential diagnosis. Technological innovations making use of both structural and functional neuroimaging methods, as well molecular biology and molecular genetic techniques, have presented in the recent literature a strong perspective for the early diagnosis of dementia, especially of Alzheimer disease. The different etiologies involved in the development of dementia syndromes, as well as the respective diagnostic conduct, will be reviewed in this article.
Dementia; differential diagnosis; Alzheimer's disease; vascular dementia; non-Alzheimer dementia
ARTIGO ORIGINAL
Diagnóstico diferencial das demências
The differential diagnosis of dementia
José Gallucci NetoI; Melissa Garcia TameliniI; Orestes Vicente ForlenzaII
IMédico psiquiatra e pesquisador do Laboratório de Neurociências (LIM-27) do Instituto de Psiquiatria do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
IIMédico psiquiatra. Doutor em medicina pelo Departamento de Psiquiatria da Universidade de São Paulo. Médico pesquisador e coordenador do Ambulatório de Transtornos da Memória do LIM-27, Instituto de Psiquiatria do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
Endereço para correspondência Endereço para correspondência Orestes V. Forlenza Laboratório de Neurociências LIM 27, Instituto de Psiquiatria do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo Av. Dr. Ovídio Pires de Campos s/n 05403-010 São Paulo SP E-mail: forlenza@usp.br
RESUMO
As síndromes demenciais são caracterizadas pela presença de déficit progressivo na função cognitiva, com maior ênfase na perda de memória, e interferência nas atividades sociais e ocupacionais. O diagnóstico diferencial deve, primeiramente, identificar os quadros potencialmente reversíveis, de etiologias diversas, tais como alterações metabólicas, intoxicações, infecções, deficiências nutricionais etc. Nas demências degenerativas primárias e nas formas seqüelares, o diagnóstico etiológico carrega implicações terapêuticas e prognósticas.
Sabe-se que o diagnóstico definitivo da maioria das síndromes demenciais depende do exame neuropatológico. Entretanto, uma avaliação clínica cuidadosa incluindo anamnese detalhada, exames físico e neurológico, associado a determinações bioquímicas e de neuroimagem, podem possibilitar maior acurácia no diagnóstico diferencial. Inovações tecnológicas servindo-se de métodos de neuroimagem estrutural e funcional, bem como de técnicas de biologia e genética molecular, têm apresentado perspectivas para o diagnóstico precoce das demências, particularmente da doença de Alzheimer. As diversas etiologias implicadas no desenvolvimento de síndromes demenciais, bem como as respectivas condutas diagnósticas, serão revistas neste artigo.
Palavras-chave: Demência, diagnóstico diferencial, doença de Alzheimer, demência vascular, demência não-Alzheimer.
ABSTRACT
Dementia is a syndrome characterized by progressive impairment of cognitive functions, particularly in memory, which affects social and occupational activities. The differential diagnosis must, firstly, identify potentially treatable causes of cognitive impairment, addressing the different etiologies of reversible dementia such as metabolic alterations, intoxications, CNS infections, and nutritional deficiencies. The correct and early diagnosis of primary degenerative dementia carries therapeutic and prognostic implications, which may attenuate the inevitable cognitive and behavioral deficits.
Definitive diagnoses of most primary dementia syndromes rely on post-mortem neuropathological examination. However, a thorough clinical evaluation, including a detailed clinical history, physical and neurological examination, combined with biochemical determinations and neuroimaging, provide a more accurate differential diagnosis. Technological innovations making use of both structural and functional neuroimaging methods, as well molecular biology and molecular genetic techniques, have presented in the recent literature a strong perspective for the early diagnosis of dementia, especially of Alzheimer disease. The different etiologies involved in the development of dementia syndromes, as well as the respective diagnostic conduct, will be reviewed in this article.
Key words: Dementia, differential diagnosis, Alzheimer's disease, vascular dementia, non-Alzheimer dementia.
Doença de Alzheimer
A doença de Alzheimer (DA) responde por cerca de 60% de todas as demências, o que a torna a causa principal de demência (LoGiudice, 2002). Quanto ao curso clínico, apresenta-se com início insidioso e deterioração progressiva. O prejuízo de memória é o evento clínico de maior magnitude. Nos estágios iniciais, geralmente encontramos perda de memória episódica e dificuldades na aquisição de novas habilidades, evoluindo gradualmente com prejuízos em outras funções cognitivas, tais como julgamento, cálculo, raciocínio abstrato e habilidades visuo-espaciais. Nos estágios intermediários, pode ocorrer afasia fluente, apresentando-se como dificuldade para nomear objetos ou para escolher a palavra adequada para expressar uma idéia, e também apraxia. Nos estágios terminais, encontram-se marcantes alterações do ciclo sonovigília; alterações comportamentais, como irritabilidade e agressividade; sintomas psicóticos; incapacidade de deambular, falar e realizar cuidados pessoais.
O diagnóstico definitivo de DA só pode ser feito mediante a análise histopatológica do tecido cerebral post-mortem. As alterações histopatológicas incluem perda neuronal nas camadas piramidais do córtex cerebral e degenerações sinápticas intensas, tanto em nível hipocampal quanto neocortical (Braak e Braak, 1991). Entretanto, a DA caracteriza-se histopatologicamente por duas lesões principais, as placas senis que contém a proteína b-amilóide, e os emaranhados neurofibrilares (revisão sobre o assunto por Caramelli, 2000). Os critérios diagnósticos para a DA segundo o NINCDS-ADRDA (National Institute for Communicative Disorders and StrokeAlzheimer's Disease and Related Disorders Association), de McKhann et al. (1984) encontram-se no item 1 do apêndiceapêndice.
A DA tem etiologia ainda desconhecida, excetuando-se os raros casos familiares, de início precoce, nos quais encontra-se mutação genética específica. Os fatores genéticos parecem ser muito relevantes, sabe-se que uma história familiar positiva para DA é o único fator sistemático associado à doença, excetuando-se a idade (Heyman, 1984). A DA pode ser transmitida de forma autossômica dominante, e as características de idade de início e evolução são determinadas pelos diferentes subtipos genéticos. Os defeitos genéticos localizados nos cromossomos 14 e 21 estão relacionados a formas de início precoce (abaixo dos 65 anos) da doença. Os cromossomos implicados, até o momento, nos subtipos genéticos são o 14 (gene PS-1), 21 (gene APP), 1 (gene PS-2) e 19 (apoE e4/e4, apoE e3/e3) (Goate 1991; Levy-Lahad, 1995; Van Duijn 1996).
Não há marcadores específicos na investigação laboratorial e de imagem da DA, embora alguns achados possam dar suporte ao diagnóstico clínico. Os exames subsidiários convencionais na DA permitem não apenas a exclusão de causas reversíveis, como também a detecção de parâmetros de neuroimagem compatíveis com os diferentes estágios clínicos da doença, tais como atrofia cortical, alterações hipocampais e temporais mesiais (revisão por Poulin e Zakzanis 2002). Em nível experimental, marcadores moleculares da doença têm sido pesquisados em fluidos biológicos, particularmente no líquido cefalorraqueano. Como exemplo, podemos citar a determinação dos títulos do peptídeo b-amilóide (e suas frações) e da proteína tau hiperfosforilada por métodos que variam do imunodiagnóstico (Höglund et al., 2004; Hampel et al., 2004; Andreassen et al., 2003) à espectrometria de massa (Lewczuk et al., 2004). Alguns parâmetros de neuroimagem associados a novas tecnologias também podem, em futuro próximo, contribuir para o diagnóstico precoce da DA. Entre essas perspectivas, podemos citar a determinação volumétrica seqüencial de regiões cerebrais de interesse, pela ressonância magnética estrutural (Pennanen et al., 2004), ou, conforme recentemente apresentado, o desenvolvimento de traçadores radioativos para PET capazes de se ligar especificamente aos depósitos cerebrais de b-amilóide (Klunk et al., 2004). Marcadores genéticos ainda não têm finalidade diagnóstica, exceto na determinação de mutações específicas associadas a formas raras da doença. As alterações genotípicas, tais como a presença de apolipoproteína e4 e outros polimorfismos, que contribuem para a avaliação do risco de doença, também não têm, no momento, indicação diagnóstica. Novas perspectivas laboratoriais, utilizando-se de técnicas de genética molecular e análise de expressão gênica, também têm proposto subsídios ao diagnóstico da DA (Blalock et al., 2004).
Demência vascular
É largamente aceito que as doenças cerebrovasculares possam ser responsáveis pelo desenvolvimento de quadros demenciais. As demências vasculares (DV) constituem a segunda maior causa de demência. Entretanto ainda não há consenso sobre os mecanismos fisiopatológicos exatos que levam à demência (Román, 2002).
A definição de demência classicamente prioriza o comprometimento de memória (por acometimento cortical, tal como na DA) (APA, 2000a); entretanto, falha na identificação de grande número de pacientes, cujo comprometimento primário ocorre em outros domínios cognitivos. Os pacientes com DV tipicamente apresentam-se com síndrome demencial do tipo córtico-subcortical, na qual os sintomas primários são de déficits nas funções executivas ou focais múltiplos (Román, 2002).
A apresentação clínica da DV depende da causa e localização do infarto cerebral (Rockwood et al., 1999). Doença de grandes vasos leva comumente a múltiplos infartos corticais (com síndrome demencial cortical multifocal), enquanto uma doença de pequenos vasos, geralmente resultado de hipertensão arterial sistêmica (HAS) e diabetes melito, causa isquemia da substância branca periventricular e infartos lacunares (com demência subcortical, alterações frontais, disfunção executiva, comprometimento de memória, prejuízo atencional, alterações depressivas, lentificação motora, sintomas parkinsonianos, distúrbios urinários e paralisia pseudobulbar) (Román, 2002).
Em estudo recente (Groves et al., 2000), poucas diferenças consistentes foram identificadas entre DA e DV. Pacientes com DV mostraram taxas mais elevadas de depressão e comprometimento funcional, além de menor comprometimento cognitivo quando comparados a pacientes com DA. Duas classificações têm sido usadas para o diagnóstico de DV: o critério desenvolvido pelo NINDS-AIREN (National Institute of Neurological Disorders and Stroke and the Association Internationale pour la Recherche et l'Ènseignement en Neurosciences) e o critério do CAD-DTC (California Alzheimer's Disease Diagnostic and Treatment Centers). Para ambos os critérios, três elementos são fundamentais: síndrome demencial, doença cerebrovascular e relação temporal razoável entre ambas. Os critérios diagnósticos para demência vascular segundo NINDS-AIREN (Roman et al., 1993) encontram-se no item 2 do apêndiceapêndice, e as principais diferenças entre as duas classificações encontram-se na Tabela 1.
Na prática clínica, o escore isquêmico de Hachisnki também providencia elementos adicionais para o diagnóstico da DV. Escore maior ou igual a 7 é consistente com DV, escore menor ou igual a 4 com DA e escore de 5 a 6, é sugestivo de DA associada a DV. Em metanálise recente, os eventos mais freqüentemente encontrados em DV que em DA são os seguintes: deterioração em "degraus", curso flutuante, história prévia de HAS ou acidente vascular cerebral (AVC), além da presença de déficits neurológicos focais (Moroney et al., 1997).
Demência mista
A demência mista é entidade nosológica caracterizada pela ocorrência simultânea de eventos característicos de DA e DV. De acordo com estudos patológicos, estima-se que mais de um terço dos pacientes com DA apresentem também lesões vasculares, e proporção similar de pacientes com DV exibam alterações patológicas características de DA (Kalaria e Ballard, 1999).
A presença de lesões vasculares nos pacientes com DA pode estar subestimada e parece estar associada à deterioração clínica mais rápida (Román 2002, Pasquier et al. 1997; Snowdon et al., 1997; Nagy et al.,1997). A apresentação mais comum de demência mista é a de um paciente com sintomas e características clínicas típicas de DA que sofre piora abrupta, acompanhada pela presença de sinais clínicos de AVC. Esta forma de demência mista foi chamada de demência pré-AVC por Hénon et al. (2001) e sua detecção pode ser auxiliada pelo uso do IQCODE, entrevista com parentes ou cuidadores.
Apesar do número de similaridades entre DA e DV, tem relativamente função superior a memória verbal de longo prazo e mais comprometimento da função frontal executiva (Tierney et al., 2001).
Demência por corpúsculos de Lewy
A demência por corpúsculos de Lewy (DCL) acomete cerca de 20% dos pacientes com demência. O diagnóstico clínico é feito quando o declínio cognitivo é flutuante, acompanhado por alucinações visuais e sintomas extrapiramidais. O quadro demencial apresenta-se com rápido início e declínio progressivo, com déficits proeminentes na função executiva, resolução de problemas, fluência verbal e performance audiovisual. As alucinações visuais são os únicos sintomas psicóticos que diferenciam DCL de DA ou DV. Quanto aos sintomas parkinsonianos, encontra-se hipomimia, bradicinesia, rigidez e, menos comumente, tremor de repouso. Sensibilidade a neurolépticos, quedas e síncopes também estão presentes (LoGiudice, 2002). A definição exclui casos em que o parkinsonismo precede a síndrome demencial em mais de 12 meses. Este critério operacional pretende excluir pacientes com doença de Parkinson que se tornaram demenciados depois, mas a praticidade deste critério diagnóstico é questionável (Galvin, 2003). Os critérios diagnósticos para demência de Lewy encontram-se no item 3 do apêndiceapêndice (McKeith et al., 1996).
Os eventos patológicos marcantes são os corpúsculos de Lewy, que são inclusões intracitoplasmáticas eosinofílicas hialinas, encontrados geralmente no córtex cerebral e no tronco encefálico (Byrne, 2000), neocorticais e uma quantidade variável de eventos patológicos relacionados com DA, como placas senis e em menor extensão emaranhados neurofibrilares (Galvin, 2003).
A presença de declínio cognitivo associado a parkinsonismo, ataxia e instabilidade autonômica com início gradual ao redor da quinta ou sexta década de vida deve levantar a suspeita de atrofia de múltiplos sistemas. Clinicamente, a atrofia de múltiplos sistemas tem apresentação variada, de acordo com a área do sistema nervoso central afetada (Papp e Lantos, 1994). Distinguem-se três apresentações clínicas, mas a sobreposição entre elas é comum. A variante estriatonigral apresenta-se com início gradual de parkinsonismo, hiper-reflexia, mioclonias, ataxia e insuficiência autonômica. A variante olivo-pontocerebelar apresenta-se com parkinsonismo leve, disartria, fala escandida e instabilidade autonômica. Raramente, esta variante pode ser acompanhada de mudanças na personalidade e demência (Critchley e Greenfield, 1948). A variante Shy-Drager (Shy e Drager, 1960) é caracterizada por falência autonômica, com incontinência ou retenção urinária, síncopes, impotência, e, menos freqüentemente, incontinência fecal. Uma história associada de distúrbio do comportamento do sono REM pode, com freqüência, auxiliar o diagnóstico de atrofia de múltiplos sistemas (Wenning et al., 1994).
Demência frontotemporal
O termo demência frontotemporal (DFT) caracteriza uma síndrome neuropsicológica marcada por disfunção dos lobos frontais e temporais, geralmente associada à atrofia dessas estruturas, e relativa preservação das regiões cerebrais posteriores. Estima-se que a DFT responda por 10% a 15% dos casos de demência degenerativa, ocorrendo principalmente após os 40 anos de idade, com igual incidência em homens e mulheres (Mendez et al., 1993).
Os achados histopatológicos típicos da DFT são consideravelmente heterogêneos e a presença dos corpúsculos de Pick não é patognomônica. Embora a DFT e a DA só possam ser definitivamente diagnosticadas através do estudo histopatológico, algumas características clínicas distinguem as duas síndromes durante a vida.
Os déficits mais característicos da DA envolvem a memória episódica, refletindo o prejuízo funcional nas áreas cerebrais mais susceptíveis à patologia da DA (lobo temporal médio). Na medida em que há progressão para outras regiões cerebrais, os sintomas passam a envolver outros déficits cognitivos, sociais e comportamentais. A DFT, entretanto, tem início seletivo nos lobos frontais e temporais anteriores e os pacientes nos estágios iniciais da doença mostram discreto comprometimento da memória episódica, mas exibem importantes alterações comportamentais (Rosen et al., 2002). Tais alterações incluem mudanças precoces na conduta social, desinibição, rigidez e inflexibilidade mentais, hiperoralidade, comportamento estereotipado e perseverante, exploração incontida de objetos no ambiente, distraibilidade, impulsividade, falta de persistência e perda precoce da crítica. O início dos sintomas antes dos 65 anos de idade, história familiar positiva em parentes de primeiro grau e a presença de paralisia bulbar, acinesia, fraqueza muscular e fasciculações (doença do neurônio motor) dão suporte ao diagnóstico. Na DFT os prejuízos cognitivos começam tipicamente nas funções executivas (Perry e Hodges, 2000), mas podem também envolver a linguagem. As várias características clínicas associadas à DFT raramente são vistas em pacientes com DA. Baseado nestas características, em 1998, desenvolveu-se consenso para o diagnóstico clínico da DFT (Neary et al., 1998), dividindo-a em três síndromes clínicas: DFT, afasia progressiva não-fluente e demência semântica. Tal classificação reflete a heterogeneidade clínica da DFT, mas está embasada nos diferentes acometimentos do lobo frontal e temporal, bem como no envolvimento distinto dos hemisférios cerebrais, e suas respectivas manifestações clínicas. O envolvimento predominante do lobo frontal está associado com alterações comportamentais, incluindo desinibição, apatia, embotamento afetivo e perda da crítica da conduta pessoal. Esta síndrome clínica continua sendo reconhecida como DFT, mas pode ser referida por alguns autores com variante frontal da DFT (Hodges e Miller, 2001). Há ainda algumas evidências que apontam a associação entre a variante frontal da DFT com atrofia frontal à direita (Fukui, e Kertesz 2000). Em contraste, o acometimento cerebral em lobo frontal esquerdo está associado com perda progressiva da fala, caracterizada por fala hesitante, dita não-fluente. Tal apresentação clínica tem sido chamada de afasia progressiva não-fluente. Já o envolvimento primordial em lobo temporal anterior esquerdo está associado com perda do conhecimento em relação a palavras e objetos, e é, portanto, conhecido como demência semântica (Hodges, 1992, Snowden, 1989). Na demência semântica, as alterações comportamentais apresentam-se de forma muito semelhante às alterações da DFT, em contrapartida, na afasia progressiva não-fluente as alterações de comportamento quase sempre estão ausentes nos estágios iniciais da doença, podendo aparecer mais tardiamente. Os critérios diagnósticos para demência frontotemporal, propostos pelos grupos de Lund e Manchester (1994) encontram-se no item 4 do apêndiceapêndice.
Doença de Huntington (DH)
A DH é doença autossômica dominante heredodegenerativa caracterizada por distúrbio do movimento, sintomas psiquiátricos e demência. É causada pela expansão do trinucleotídeo CAG no gene que codifica a proteína huntingtina, localizado no cromossomo 4 (4p16.3). A demência torna-se usualmente aparente após o surgimento dos sintomas coréicos e psiquiátricos. A memória é afetada em todos os aspectos e o aparecimento de afasia, apraxia, agnosia e disfunção cognitiva global ocorrem mais tardiamente (Quinn e Schrag, 1998).
Doença de Creutzfeldt-Jakob (DCJ)
DCJ é o protótipo de doença causada por príons em humanos. Trata-se de uma enfermidade infecciosa e invariavelmente fatal, que atinge o sistema nervoso central e caracteriza-se por demência rapidamente progressiva e envolvimento focal variável do córtex cerebral, gânglios da base, cerebelo, tronco cerebral e medula espinhal. Embora a transmissão de humanos para animais tenha sido demonstrada experimentalmente, a transmissão entre seres humanos (por transplante de córnea, implantação de eletrodos corticais etc.) parece ser rara (Johnson e Gibbs, 1998). O agente infeccioso está presente no cérebro, medula espinhal, olhos, pulmões, linfonodos, rins, baço, fígado, no líquido cefalorraqueano, mas não em outros fluídos corporais.
Demências reversíveis
As demências reversíveis são causas raras de demência. Entretanto, são importantes do ponto de vista diagnóstico, pois o tratamento adequado pode reverter o declínio cognitivo.
Hidrocefalia de pressão normal (HPN)
A HPN é causa potencialmente reversível de demência e caracteriza-se pela tríade clássica: demência, ataxia e incontinência urinária. Pode ser idiopática ou secundária a condições que interfiram na absorção liquórica, como meningite ou hemorragia subaracnóide. A demência é freqüentemente leve e de início insidioso, e é tipicamente precedida por distúrbio de marcha e incontinência urinária (Vanneste, 2000). A deterioração da memória é comum, mas o aparecimento de apraxia e agnosia é incomum.
Pelagra
Causada pela deficiência de ácido nicotínico (niacina), afeta os neurônios do córtex cerebral, gânglios da base, tronco cerebral, cerebelo e corno anterior da medula espinhal. Manifesta-se por diarréia, glossite, anemia e lesões cutâneas eritematosas. Pode produzir demência, psicose, estados confusionais, sinais cerebelares e extrapiramidais, polineuropatia e neuropatia óptica.
Deficiência de vitamina B12
A deficiência de vitamina B12 é causa rara de demência reversível e psicose orgânica. A demência é caracterizada por disfunção cognitiva global, lentificação mental, perda de memória e dificuldade de concentração. Déficits corticais focais não ocorrem. As manifestações psiquiátricas são proeminentes e incluem depressão, mania e quadros psicóticos com alucinações auditivas e visuais (Toh et al., 1997).
Hipotireoidismo
O hipotireoidismo é causa de demência reversível e psicose crônica. A demência é caracterizada por lentificação mental, perda de memória e irritabilidade. Déficits corticais focais não ocorrem. As manifestações psiquiátricas são proeminentes e incluem depressão, paranóia, alucinações auditivas e visuais, mania e comportamento suicida.
Depressão
A depressão é o quadro que gera maior confusão diagnóstica com demência. Como a depressão é condição potencialmente tratável, a distinção entre as duas condições torna-se obrigatória. Tanto a depressão quanto a demência causam lentificação psíquica, apatia, irritabilidade, descuido pessoal, dificuldades com concentração e memória, e mudanças no comportamento e personalidade. Além disto, a depressão pode ser um sintoma da demência e, não raramente, ambas as situações coexistem (Raskind, 1998). Na tabela 2, as características clínicas que auxiliam no diagnóstico diferencial entre depressão e demência.
Demências infecciosas
Complexo aids-demência
O complexo aids-demência é a complicação neurológica mais comum da síndrome da imunodeficiência adquirida. Ocorre mais freqüentemente em pacientes gravemente imunocomprometidos, em estágios avançados da doença. Entretanto, pode ser uma das apresentações iniciais da aids (Simpson e Tagliati, 1994). A demência é de início subagudo e caracteriza-se por apatia, dificuldade de concentração, prejuízo de memória verbal e funções executivas, disfunção motora e distúrbios de comportamento. Com a progressão pode haver ataxia, disartria, mutismo, delírios e alucinações visuais (APA, 2000b).
Neurossífilis
Era causa freqüente de demência antes do aparecimento e disseminação do uso da penicilina no tratamento das fases iniciais da doença. Embora incomum nos dias de hoje, a demência por neurossífilis ainda constitui diagnóstico diferencial a considerar-se diante de síndromes demenciais atípicas ou com manifestações frontais, particularmente em populações menos favorecidas socialmente (Simon, 1994).
Encefalite herpética
Embora incomum, permanece como a principal causa de encefalite esporádica na América do Norte. Quando não tratada precocemente tem prognóstico desfavorável. Cerca de metade dos pacientes morre em dias ou poucas semanas. Dos que sobrevivem, a maioria tem seqüelas significativas, tais como as síndromes de Korsakoff e Kluver-Bucy, sinais neurológicos focais como hemiparesia e afasia, tiques vocais e motores, e demência (McGrath et al., 1997).
Neurocisticercose
Em nosso meio, a neurocisticercose é considerada doença endêmica e pode levar a comprometimento cognitivo por inflamação crônica das meninges, hidrocefalia obstrutiva e pela presença de cistos corticais. O quadro clínico resulta do próprio processo inflamatório, do número e localização das lesões, além de prejuízos secundários a infecções prévias (Sotelo et al., 1985). São comuns manifestações típicas da demência vascular (Alarcón et al., 1992).
Alcoolismo
Algumas complicações do alcoolismo podem causar demência. Estas incluem o hematoma subdural crônico por traumatismo craniano, a degeneração hepatocerebral por cirrose hepática e deficiências nutricionais. A demência como acometimento neurológico por efeito tóxico direto do álcool tem sido proposta, embora nenhuma anormalidade distintiva tenha sido observada nos cérebros dos pacientes com demência pelo uso do álcool (Victor, 1994).
Síndrome de Wernicke-Korsakoff
A síndrome de Wernicke ocorre pela deficiência de tiamina associada ao uso crônico de álcool, e é transtorno neurológico agudo caracterizado por ataxia, disfunção vestibular, delírio e pela variedade de anormalidades da motricidade ocular (nistagmo, paralisia dos retos laterais e paralisia da fixação). Se não tratada adequadamente pode evoluir para síndrome amnéstica crônica conhecida com síndrome de Korsakoff, onde os aspectos essenciais são o prejuízo grave de memória recente e aprendizado (amnésia anterógrada) e confabulações, com pobre resposta ao tratamento.
Outras etiologias
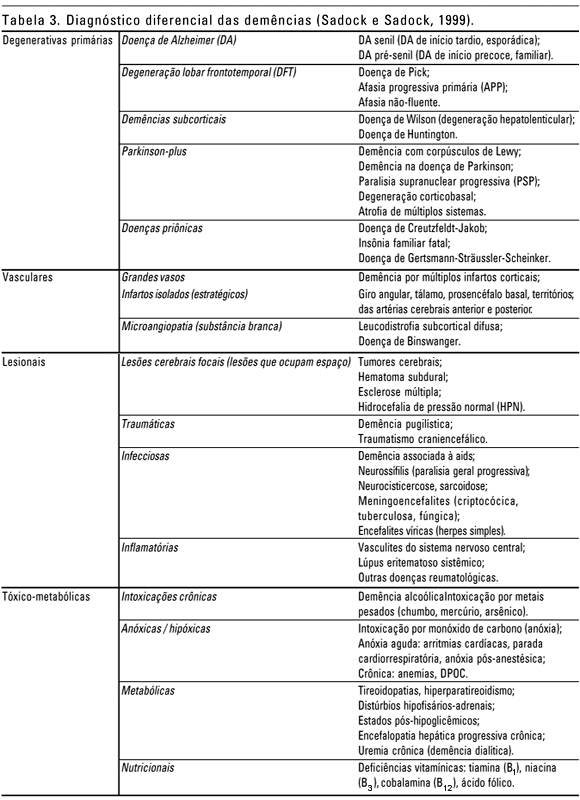
Diversas outras condições médicas estão associadas com a presença de sintomas demenciais. Elas incluem diversos distúrbios metabólicos e uso de medicações (principalmente com efeitos anticolinérgicos, analgésicos opiáceos e esteróides adrenocorticias). A tabela 3 reúne as diferentes etiologias para as síndromes demenciais.
Recebido: 23/03/2005 - Aceito: 28/04/2005
1) Doença de Alzheimer:
Doença de Alzheimer PROVÁVEL segundo critérios do NINCDS-ADRDA (McKhann et al., 1984)
I. Critérios para o diagnóstico clínico de doença de Alzheimer PROVÁVEL:
-
Demência estabelecida por exame clínico e documentada pelo Mini-Exame do Estado Mental, escala de demência de Blessed, ou avaliação similar, e confirmada por testes neuropsicológicos;
-
Déficits em duas ou mais áreas da cognição;
-
Piora progressiva da memória e outras funções cognitivas;
-
Ausência de distúrbio da consciência;
-
Início entre os 40 e 90 anos, mais freqüentemente após os 65 anos; e
-
Ausência de doenças sistêmicas ou outras doenças cerebrais que por si só possam provocar declínio progressivo de memória e cognição.
II. O diagnóstico de doença de Alzheimer PROVÁVEL é auxiliado por:
-
Deterioração progressiva de funções cognitivas específicas como linguagem (afasia), habilidade motora (apraxia) e percepção (agnosia);
-
Prejuízo nas atividades do dia-a-dia e padrões anormais de comportamento;
-
História familiar de demência (particularmente se confirmada por exame neuropatológico);
-
Exames laboratoriais compatíveis com o diagnóstico:
· punção lombar: normal, pelas técnicas usuais;
· EEG: padrão normal ou alterações inespecíficas, como aumento de ondas lentas;
· TC de crânio: atrofia cerebral, com progressão documentada por exames seriados.
III. Outras características clínicas consistentes com o diagnóstico de doença de Alzheimer PROVÁVEL, após exclusão de outras causas de demência, incluem:
-
"Platôs" no curso progressivo da doença;
-
Sintomas associados: depressão, insônia, incontinência, delírios, ilusões, alucinações, incontinência verbal, explosões emocionais, agitação, distúrbios sexuais, perda de peso;
-
Outras anormalidades neurológicas, observadas em alguns pacientes, especialmente com doença avançada e incluindo sinais motores, aumento de tônus muscular, mioclonias ou distúrbio da marcha;
-
Convulsões em doença avançada;
-
TC de crânio normal para a idade.
IV. As características que tornam o diagnóstico de doença de Alzheimer PROVÁVEL incerto ou pouco provável incluem:
-
Início abrupto;
-
Sinais neurológicos focais (tais como hemiparesia, déficits sensitivos, déficits em campos visuais, distúrbio da coordenação motora) no início do curso da doença; e
-
Convulsões ou distúrbios da marcha nos estágios iniciais da doença.
2) Demência vascular
Demência vascular provável segundo critérios do NINDS-AIREN (Roman et al., 1993)
I. Os critérios para o diagnóstico clínico de demência vascular PROVÁVEL incluem todos os seguintes:
1. Demência, definida por declínio cognitivo a partir de um funcionamento prévio superior ao nível atual e manifestada por prejuízo de memória e de dois ou mais domínios cognitivos (orientação, atenção, linguagem, funções visoespaciais, funções executivas, controle motor e praxia), preferencialmente estabelecida por avaliação clínica e documentada por testes neuropsicológicos; os déficits devem ser graves o suficiente para causar prejuízos nas atividades do dia-a-dia, e não devidos à conseqüência física de um acidente vascular cerebral (AVC) isolado.
Critérios exclusão: rebaixamento do nível de consciência, delirium, psicose, afasia ou prejuízo sensoriomotor importantes. Também excluem o diagnóstico a presença de doenças sistêmicas outras que não-cerebrais, que por si só podem ser responsáveis pelo declínio cognitivo e memória.
2. Doença cerebrovascular, definida pela presença de sinais focais ao exame neurológico, tais como hemiparesia, hipotonia facial, sinal de Babinski, déficit sensorial, hemianopsia, e disartria consistentes com lesão vascular (com ou sem história de AVC), e evidência de doença cerebrovascular relevante em exame de imagem (tomografia computadorizada ou ressonância magnética) incluído infartos múltiplos de grandes vasos ou infarto único estrategicamente localizado (giro angular, tálamo, prosencéfalo basal ou nos territórios da artéria cerebral anterior e posterior), assim como múltiplas lacunas em gânglios da base e substância branca ou lesões extensas em substância branca periventricular, ou uma combinação de ambas.
3. Uma relação entre os distúrbios citados acima, manifestada ou inferida pela presença de um ou mais dos seguintes: (a) início da demência dentro de três meses após um AVC reconhecido; (b) deterioração abrupta das funções cognitivas; ou flutuações, progressão em "degraus" dos déficits cognitivos.
II. Características clínicas compatíveis com o diagnóstico de demência vascular PROVÁVEL:
-
Presença precoce de distúrbio da marcha (marcha em
petits pas, ou robótica, apráxica-atáxica ou marcha parkinsoniana);
-
História de desequilíbrio e freqüentes quedas não provocadas;
-
Urgência urinária precoce e outros sintomas urinários não explicados por doença urológica;
-
Paralisia pseudobulbar;
-
Mudanças na personalidade e no humor, abulia, depressão, incontinência emocional, ou outros déficits subcorticais, incluindo retardo psicomotor e função executiva anormal.
III. Características que tornam o diagnóstico de demência vascular incerto ou pouco provável:
-
Início precoce de déficit mnéstico e piora progressiva da memória e outras funções cognitivas como linguagem (afasia sensorial transcortical), habilidade motora (apraxia) e percepção (agnosia), na ausência de lesões focais correspondentes em exame de imagem;
-
Ausência de sinais neurológicos focais, além do distúrbio cognitivo; e
-
Ausência de lesões cerebrovasculares em exames de imagem (TC ou RNM).
3) Demência com corpúsculos de Lewy:
Demência provável por corpúsculos de Lewy (DCL) segundo critérios de McKeith et al. (1996).
1. A característica central necessária para o diagnóstico de DCL é um declínio cognitivo progressivo de magnitude suficiente para interferir com a função social ou ocupacional. Prejuízo proeminente ou persistente de memória pode não ocorrer necessariamente nos estágios iniciais, mas é normalmente evidente com a progressão do quadro. Déficits atencionais e de habilidades fronto-subcorticais e visoespaciais podem ser proeminentes.
2. Duas das seguintes características centrais são essenciais para o diagnóstico de DCL PROVÁVEL:
a) Cognição flutuante, com pronunciada variação na atenção e no estado de alerta;
b) Alucinações visuais recorrentes que são tipicamente bem formadas e detalhadas;
c) Características motoras espontâneas de parkinsonismo.
3. Características que dão suporte ao diagnóstico são
-
Quedas repetidas;
-
Síncope;
-
Perda transitória da consciência;
-
Sensibilidade a neurolépticos;
-
Delírios sistematizados;
-
Alucinações em outras modalidades.
4. O diagnóstico de DCL é menos provável na presença de:
-
Acidente vascular cerebral, evidenciado por sinal neurológico focal ou exame de imagem;
-
Evidência no exame físico e investigação de qualquer doença física ou outra doença cerebral responsável pelo quadro clínico.
4) Demência frontotemporal:
Critérios propostos pelos grupos de Lund e Manchester (1994):
I. Características diagnósticas centrais:
1) Distúrbios do comportamento:
-
Início insidioso e progressão lenta;
-
Perda precoce da consciência pessoal (negligência com higiene pessoal);
-
Perda precoce da consciência social (ausência de tato social, desvios da conduta social);
-
Sinais precoces de desinibição (como hipersexualidade, comportamento violento, jocosidade);
-
Rigidez mental e inflexibilidade;
-
Hiperoralidade (mudanças na dieta, excessos alimentares, consumo excessivo de álcool e tabaco, exploração oral dos objetos);
-
Comportamento esterotipado e perseverativo (comportamento errante, maneirismos como bater palmas, cantar, dançar, preocupação ritualística como colecionismo, hábitos de higiene íntima e vestuário);
-
Comportamento de utilização (exploração irrestrita de objetos no ambiente);
-
Distratibilidade, impulsividade e falta de persistência;
-
Perda precoce do
insight e da crítica de que as alterações são secundárias a uma alteração no próprio estado mental.
2) Sintomas afetivos:
-
Depressão, ansiedade, sentimentalismo excessivo, ideação suicida, delírios (presentes precocemente e efêmeros);
-
Hipocondria, preocupação somática bizarra (precoces e efêmeros);
-
Indiferença emocional (ausência de empatia e simpatia, apatia);
-
Perda da mímica (inércia e falta de espontaneidade).
3) Distúrbio da fala:
-
Redução progressiva da fala (redução das manifestações verbais espontâneas);
-
Fala estereotipada (repetição de repertório limitado de palavras, frases e temas);
-
Ecolalia e perseveração;
-
Mutismo tardio.
4) Orientação espacial e praxia preservadas (habilidades intactas para interagir com o ambiente).
5) Sintomas físicos:
-
Reflexos primitivos precoces;
-
Incontinência precoce;
-
Acinesia (tardia), rigidez, tremor;
-
Pressão sangüínea baixa e lábil.
6) Investigações complementares:
-
EEG normal apesar de demência clinicamente evidente;
-
Exame de imagem (estrutural ou funcional, ou ambos) evidenciando anormalidade predominantemente frontal ou temporal anterior, ou frontotemporal;
-
Exame neuropsicológico alterado (graves prejuízos nos testes dos lobos frontais, na ausência de amnésia significativa, afasia, ou distúrbio espacial).
II. Características que dão suporte ao diagnóstico:
-
Início antes dos 65 anos;
-
História familiar positiva de distúrbio similar em parente de primeiro grau;
-
Paralisia bulbar, fraqueza muscular, fasciculações (doença do neurônio motor).
III. Características incompatíveis com o diagnóstico (critérios de exclusão):
-
Início abrupto com eventos ictais;
-
Trauma de crânio relacionado ao início dos sintomas;
-
Amnésia precoce e importante;
-
Desorientação espacial precoce, desorientação em ambientes familiares, dificuldades para localizar objetos;
-
Apraxia precoce e importante;
-
Fala logoclônica, com perda rápida da associação das idéias;
-
Mioclonias;
-
Déficit cortical bulbar ou medular;
-
Ataxia cerebelar;
-
Coreoatetose;
-
EEG significativamente anormal em estágios iniciais da doença;
-
Exame de imagem com alterações estruturais ou funcionais predominantemente em estruturas pós-centrais; lesões multifocais na TC ou RNM de crânio;
-
Testes laboratoriais indicando envolvimento cerebral ou doença inflamatória (como esclerose múltipla, sífilis, AIDS e encefalite herpética).
IV. Características pouco compatíveis com diagnóstico:
-
História típica de alcoolismo crônico;
-
Hipertensão sustentada;
-
História de doença vascular (como angina, claudicação).
- ALARCÓN, F.; HIDALGO, F.; MONCAYO, J. et al. - Cerebral Cysticercosis and Stroke. Stroke 23:224-8, 1992.
- AMERICAN PSYCHIATRIC ASSOCIATION (APA) - Dementia. In: DSM-IV. 4thed. Washington. DC: American Psychiatric Association, pp.147-71, 2000a.
- AMERICAN PSYCHIATRIC ASSOCIATION (APA) - Practice Guideline for Treatment of Patients With HIV Washington. DC: American Psychiatric Association, pp.14-7, 2000b.
- ANDREASEN, N.; VANMECHELEN, E.; VANDERSTICHELE, H. et al. - Cerebrospinal Fluid Levels of Total-Tau, Phospho-Tau and A Beta 42 Predicts Development of Alzheimer's Disease in Patients with Mild Cognitive Impairment. Acta Neurol Scand 179:47-51, 2003.
- BLALOCK, E.M.; GEDDES, JW.; CHEN, K.C. et al. - Incipient Alzheimer's Disease: Microarray Correlation Analyses Reveal Major Transcriptional and Tumor Suppressor Responses. Proc Natl Acad Sci USA 101(7):2173-8, 2004.
- BRAAK, H.; BRAAK, E. - Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol 82:239-59, 1991.
- BYRNE, J. - Doença de Parkinson e Demência com Corpúsculo de Lewy, In: Forlenza, O.V.; Carameli, P. (eds). Neuropsiquiatria geriátrica Rio de Janeiro: Atheneu, pp.215-20, 2000.
- CARAMELLI, P. - Neuropatologia da Doença de Alzheimer, In: Forlenza, O.V.; Carameli, P. (eds). Neuropsiquiatria geriátrica Rio de Janeiro: Atheneu, pp.107-18, 2000.
- CRITCHLEY, M.; GREENFIELD, J.G. - Olivo-Ponto-Cerebellar Atrophy. Brain 71: 343-64, 1948.
- FORLENZA, O.V.; NITRINI, R. - Doença de Alzheimer. In: Fráguas, R. Jr.; Figueiró J.A.B. (eds). Depressões Secundárias: depressões associadas a condições médicas e medicamentos. Rio de Janeiro: Atheneu, pp.109-18, 2001.
- FUKUI, T.; KERTESZ, A. - Volumetric Study of Lobar Atrophy in Pick Complex and Alzheimer's Disease. J Neurol Sci 174: 111-21, 2000.
- GALVIN, J. - Dementia with Lewy Bodies. Arch Neurol 60(9):1332-5, 2003.
- GOATE, A.; CHARTIER-HARLIN, M.C.; MULLAN, M. et at. - Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer's Disease. Nature 349(6311):704-6, 1991.
- GROVES, W.C.; BRANDT, J.; STEINBERG, M. et al. - Vascular Dementia and Alzheimer's Disease: is There a Difference? A Comparation of Symptoms by Disease Duration. J Neuropsychiatry Clin Neurosci 12:305-15, 2000.
- HAMPEL, H.; BUERGER, K.; ZINKOWSKI, R. et al. - Measurement of Phosphorylated Tau Epitopes in the Differential Diagnosis of Alzheimer Disease: a Comparative Cerebrospinal Fluid Study. Arch Gen Psychiatry 61(1):95-102, 2004.
- HOGLUND, K.; WIKLUND, O.; VANDERSTICHELE, H. et al. - Plasma Levels of Beta-Amyloid(1-40), Beta-Amyloid(1-42), and Total Beta-Amyloid Remain Unaffected in Adult Patients with Hypercholesterolemia After Treatment with Statins. Arch Neurol 61(3):333-7, 2004.
- HÉNON, H.; DURIEU, I.; GUEROUAOU, D. et al. - Post-Stroke Dementia: Incidence and Relationship to Pre-Stroke Cognitive Decline. Neurology 57: 1216-22, 2001.
- HEYMAN, A.; WILKINSON, W.E.; STAFFORD, J.A. et al. - Alzheimer's Disease: a Study of Epidemiological Aspects. Ann Neurol 15:335-41, 1984.
- HODGES, J.R.; MILLER, B. - The Classification, Genetics and Neuropathology of Frontotemporal Dementia. Introduction to the Special Topic Papers: Part I. Neurocase 7: 31-5, 2001.
- HODGES, J.R.; PATTERSON, K.; OXBURY, S.; FUNNELL, E. - Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain 115(Pt 6): 1783-806, 1992.
- JOHNSON, R.T.; GIBBS JR., C.J. - Creutzfeldt-Jakob Disease and Related Transmissible Spongiform Encephalopathies. N Engl J Med 339:1994-2004, 1998.
- KALARIA, R.N.; BALLARD, C. - Overlap Between Pathology of the Alzheimer Disease and Vascular Dementia. Alzheimer Dis Assoc Disord 13 (Suppl. 3):S115-23, 1999.
- KLUNK, W.E.; ENGLER, H.; NORDBERG A. et al. - Imaging Brain Amyloid in Alzheimer's Disease with Pittsburgh Compound-B. Ann Neurol 55(3):306-19, 2004.
- LEVY-LAHAD, E.; WIJSMAN, E.M.; NEMENS, E. et al. - A familial Alzheimer's Disease Locus on Chromossome 1. Science 269:970-2, 1995.
- LEWCZUK, P.; ESSELMANN, H.; GROEMER, T.W. et al. - Amyloid Beta Peptides in Cerebrospinal Fluid as Profiled with Surface Enhanced Laser Desorption/Ionization Time-of-Flight Mass Spectrometry: Evidence of Novel Biomarkers in Alzheimer's Disease. Biol Psychiatry 55(5):524-30, 2004.
- LOGIUDICE, D. - Dementia: an Update to Refresh your Memory. Intern Med J 32:535-40, 2002.
- (THE) LUND AND MANCHESTER GROUPS. - Clinical and Neuropathological Criteria for Frontotemporal Dementia. J Neurol Neurosurg Psychiatry 57:416-8, 1994.
- MCGRATH, N.; ANDERSON, N.E.; CROXSON, M.C.; POWELL, K.F. - Herpes Simplex Encephalitis Treated with Acyclovir: Diagnosis and Long Term Outcome. J Neurol Neurosurg Psychiatry 63:321-6, 1997.
- MCKEITH, I.G.; GALASKO, D.; KOSAKA, K. et al. - Consensus Guidelines for the Clinical and Pathologic Diagnosis of Dementia with Lewy Bodies (DLB): Report of the Consortium on DLB International Workshop. Neurology 47:1113-24, 1996.
- MCKHANN, G.; DRACHMAN, D.; FOLSTEIN, M. et al. - Clinical Diagnosis of Alzheimer's Disease: Report of the NINCDS-ADRDA Work Group Under the Auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34:939-44, 1984.
- MENDEZ, M.F.; SELWOOD, A.; MASTRI, A.R.; FREY, W.H. - Pick's Disease Versus Alzheimer's Disease: a Comparison of Clinical, Neuropsychologival, and SPECT Characteristics. Neurology 41:1374-82, 1993.
- MORONEY, J.T.; BAGIELLA, E.; DESMOND, D.W. et al. - Meta-Analysis of the Hachisnki Ischemic Score in Pathologically Verified Dementias. Neurology 49:1096-105, 1997.
- NAGY, Z.S.; ESIRI, M.M.; JOBST, K.A. et al. - The Effects of Additional Pathology on the Cognitive Deficit of Alzheimer's Disease. J Neuropathol Exp Neurol 56:165-70, 1997.
- NEARY, D.; SNOWDEN, J.S.; GUSTAFSON, L. et al. - Frontotemporal Lobar Regeneration: a Consensus on Clinical Diagnostic Criteria. Neurology 51:1546-54, 1998.
- PAPP, M.I.; LANTOS, P.I. - The distribution of Oligodendroglial Inclusions in Multiple System Atrophy and its Relevance to Clinical Symptomatology. Brain 117:235-43, 1994.
- PASQUIER, F.; LEYS, D.; SCHELTENS, P. - The Influence of Coincidental Vascular Pathology on Syntomatology and Course of Alzheimer's Disease. JAMA 277:813-7, 1997.
- PENNANEN, C.; KIVIPELTO, M.; TUOMAINEN, S. et al. - Hippocampus and Entorhinal Cortex in Mild Cognitive Impairment and Early AD. Neurobiol Aging 25(3):303-10, 2004.
- PERRY, R.J.; HODGES, J.R. - Differentiating Frontal and Temporal Variant Frontotemporal Dementia from Alzheimer's Disease. Neurology 54:2277-84, 2000.
- POULIN, P.; ZAKZANIS, K.K. - In Vivo Neuroanatomy of Alzheimer's Disease: Evidence from Structural and Functional Brain Imaging. Brain Cogn 49(2):220-5, 2002.
- QUINN, N.; SCHRAG, A. - Huntington's Disease and other Choreas. J Neurol 245:709-16, 1998.
- RASKIND, M.A. - The Clinical Interface of Depression and Dementia. J Clin Psychiatry 59(Suppl 10):9-12, 1998.
- ROCKWOOD, K.; BOWLER, J.; ERKINJUNTTI, T.; HACHINSKI, V.; WAALIN, A. - Subtypes of Vascular Dementia. Alzheimer Dis Assoc Disord 13 (Suppl. 3):S59-65, 1999.
- ROMAN, G.C.; TATEMICHI, T.K.; ERKINJUNTTI, T. et al. - Vascular Dementia: Diagnostic Criteria for Research Studies. Report of the NINDS-AIREN International Workshop. Neurology 43:250-60, 1993.
- ROMÁN, G.C. - Defining Dementia: Clinical Criteria for the Dignosis of Vascular Dementia. Acta Neurol Scand 106 (Suppl. 178):6-9, 2002.
- ROSEN, H.J.; HARTIKAINEN, K.M.; JAGUST, W. et al. - Utility of Clinical Criteria in Differentiating Frontotemporal Lobar Degeneration (FTLD) from AD. Neurology 58:1608-15, 2002.
- SADOCK, B.J.; SADOCK, V.A. - Kaplan and Sadock's Comprehensive Textbook of Psychiatry, Seventh Edition, Philadelphia: Lippincott Williams & Wilkins, 1999.
- SHY, G.M.; DRAGER, G.A. - A Neurological Syndrome Associated with Orthostatic Hypotension: a Clinico-Pathologic Study. Arch Neurol 2:511-27, 1960.
- SIMON, R.P. - Neurosyphilis. Neurology 44: 2228-30, 1994.
- SIMPSON, D.M.; TAGLIATI, M. - Neurologic Manifestations of HIV Infection. Ann Intern Med 121:769-85, 1994.
- SNOWDEN, J.S.; GOULDING, P.J.; NEARY, D. - Semantic Dementia: a Form of Circumscribed Cerebral Atrophy. Behav Neurol 2:167-82, 1989.
- Snowdon, D.A.; Greiner, L.H.; Mortimer, JA. et al. - Brain Infarction and the Clinical Expresión of Alzheimer Disease: the Nun Study. JAMA 277:813-7, 1997.
- SOTELO, J.; GUERRERO, V.; RUBIO, F. - Neurocysticercosis: a New Classification Based on Active and Inactive Forms. A study of 753 cases. Arch Intern Med 145:442-45, 1985.
- TOH, B.H.; VAN DRIEL, I.R.; GLEESON, P.A. - Pernicious Anemia. N Engl J Med 337:1441-8, 1997.
- TIERNEY, M.C.; BLACK, S.E.; SZALAI, J.P. et al. - Recognition Memory and Verbal Fluency Differentiate Probable Alzheimer Disease from Subcortical Ischemic Vascular Dementia. Arch Neurol 58:164-9, 2001.
- VAN DUIJN, C.M. - Epidemiology of the Dementias: Recent Developments and New Approaches. J Neurol Neurosurg Psychiatry 60:478-88,1996.
- VANNESTE, J.A. - Diagnosis and Management of Normal-Pressure Hydrocephalus. J Neurol 247:5-14, 2000.
- VICTOR, M. - Alcoholic Dementia. Can J Neurol Sci 21:88-99, 1994.
- WENNING, G.K.; SHLOMO, Y.B.; MEGALHÃES, M.; DANIEL, S.E.; QUINN, N.P. - Clinical Features and Natural History of Multiple System Atrophy: an Analysis of 100 Cases. Brain 117:835-45, 1994.
apêndice
Endereço para correspondência
Datas de Publicação
-
Publicação nesta coleção
12 Ago 2005 -
Data do Fascículo
Jun 2005
Histórico
-
Recebido
23 Mar 2005 -
Aceito
28 Abr 2005