Abstracts
A DNA hybridization dot-blot assay using a radioactive and a non-radioactive probe has been developed for the detection of Aujeszky's disease virus (ADV). The Bam H I 7 fragment of ADV genomic DNA was labeled by nick translation using 32P-dCTP and the 196bp polymerase chain reaction (PCR) gG glycoprotein gene amplified fragment was also labeled by nick translation but using biotin d-7-dATP. This technique provides a fast and effective means of detecting acute cases of ADV infection but it was unable to detect ADV nucleic acid sequences in trigeminal nerve ganglia of latent infected pigs and mice.
Aujeszky's disease virus; dot-blot hybridization; radioactive probe; non radioactive probe
Uma sonda radioativa e uma outra biotinilada foram produzidas para detecção do vírus da doença de Aujeszky (VDA). O fragmento de Bam H I 7 do DNA genômico do VDA foi marcado por "nick translation" empregando P32dCTP e um fragmento de 196pb, resultante de uma amplificação pela reação em cadeia pela polimerase marcado com biotina 7-dATP. Essas sondas, de uma maneira rápida e específica, prestaram-se para a detecção da infecção aguda pelo VDA. Entretanto, não se mostraram sensíveis o suficiente para detectar seqüências genômicas de VDA no gânglio trigêmio de suínos e camundongos com infecção latente.
Vírus da doença de Aujeszky; hibridização por "dot-blot"; sonda radioativa; sonda não radioativa
Development of DNA probe for detection of Aujeszky's disease virus (Desenvolvimento de uma sonda de DNA para a detecção do vírus da doença de Aujeszky)
A.L. Cândido, P.P. Barros, M. Resende* Recebido para publicação em 10 de março de 1999. *Autor para correpondência Autor para correpondência E-mail: mresende@mono.icb.ufmg.br ABSTRACT , R.B. Flaschart
Laboratório de Virologia Comparada
Instituto de Ciências Biológicas UFMG.
Caixa Postal 486
30161-970 Belo Horizonte, MG.
A DNA hybridization dot-blot assay using a radioactive and a non-radioactive probe has been developed for the detection of Aujeszky's disease virus (ADV). The Bam H I 7 fragment of ADV genomic DNA was labeled by nick translation using 32P-dCTP and the 196bp polymerase chain reaction (PCR) gG glycoprotein gene amplified fragment was also labeled by nick translation but using biotin d-7-dATP. This technique provides a fast and effective means of detecting acute cases of ADV infection but it was unable to detect ADV nucleic acid sequences in trigeminal nerve ganglia of latent infected pigs and mice.
Keywords: Aujeszky's disease virus, dot-blot hybridization, radioactive probe, non radioactive probe.
RESUMO
Uma sonda radioativa e uma outra biotinilada foram produzidas para detecção do vírus da doença de Aujeszky (VDA). O fragmento de Bam H I 7 do DNA genômico do VDA foi marcado por "nick translation" empregando P32dCTP e um fragmento de 196pb, resultante de uma amplificação pela reação em cadeia pela polimerase marcado com biotina 7-dATP. Essas sondas, de uma maneira rápida e específica, prestaram-se para a detecção da infecção aguda pelo VDA. Entretanto, não se mostraram sensíveis o suficiente para detectar seqüências genômicas de VDA no gânglio trigêmio de suínos e camundongos com infecção latente.
Palavras-Chave: Vírus da doença de Aujeszky, hibridização por "dot-blot", sonda radioativa, sonda não radioativa.
INTRODUCTION
Aujeszky's disease virus (ADV) is the causative agent of disease in pigs, weaners and fatteners in which the mortality rate may vary widely, depending upon the age of the swine affected (Gustafson, 1986). Infection of pregnant sows may result in abortion and stillbirths. Although pigs are considered to be the host of ADV a variety of animals may also be infected and in most non swine animals the disease is usually fatal (Baskerville et al., 1973).
Like other alphaherpesvirus, a latent infection is established following the acute phase of the disease (Carneiro & Cardim, 1947; Kojnok, 1965).
To diagnose acute ADV infection the palatine tonsil and trigeminal nerve ganglia are tested for the presence of virus by either fluorescent antibody technique, cell culture, blot hybridization or PCR. Determining the occurrence of latent ADV infections requires a sensitive assay system (Maes et al., 1988; Rziha et al, 1988; White et al., 1996).
The objective of the study reported here was to develop nucleic acid hybridization probes from the ADV genome with sufficient sensitivity and specificity to aid in diagnosis of ADV infection. We selected the trigeminal ganglia as the only tissue for use in the determination of the ADV infection status of an animal.
MATERIAL AND METHODS
The ASB Piau strain of ADV was isolated from a piglet tonsil from a herd in which Aujeszky's disease (AD) was not clinically observed (Prof. Antônio Stockler Barbosa). This strain was produced in a mycoplasma free swine kidney cell line (SK-6). Cells were grown in 75cm2 flasks containing Dulbecco's Modified Eagle Medium with 10% (v/v) fetal bovine serum, 29.2mg of L-glutamine/100ml, 11mg of sodium pyruvate, 15 mM of hepes, 50mg of gentamicin/ml and 2.5mg of amphotericin B/ml. Nearly confluent monolayer of cells were infected at multiplicity of infection (MOI) of 0.01 TCID50 /cell. When cytopathic effects reached approximately 95% the virus containing medium was clarified by low speed centrifugation. The virus titer in the clarified medium was assayed by use of standard plaque counting methods (Lennette & Schmidt, 1979).
For viral DNA preparation the virions were pelleted from the cell-free culture medium by centrifugation at 35,000´g for 90min. The virus pellets were suspended in TEN (10mM Tris-HCl, pH 8,0; 1mM EDTA; 10mM NaCl) and layered onto 10% to 50% (w/w in TEN) sucrose gradients. After centrifugation at 58,700´g for 1 hour, light scattering bands at approximately the middle of tube were collected. The banded virions were pelleted and the DNA was isolated by the same treatment as described below for trigeminal nerve ganglia DNA extraction.
Three crossbred pigs with negative results of ELISA (Carvalho et al., 1994) for ADV were exposed to 2´105 TCID50 of the ASB Piau strain of ADV in each nostril at 50 days of age. Clinical signs were recorded and animals were kept in isolation for 350 days post inoculation. Pigs were euthanized by IV injection of an overdose of sodium thiopental and the trigeminal nerve ganglia was collected and placed on ice in TEN. In another set of experiments, one group (A) of mice received by the subcutaneous route 0.1ml of ultraviolet inactivated ADV (titer ³107 TCID50 /ml) and the same kind of inoculation was done three weeks later. Finally, four weeks later 0.1ml of virulent virus was given. Group (B) mice received, in the same fashion, two doses of phosphate-buffered saline and one last dose of virulent virus four weeks later. Mice were examined daily after inoculum of virulent virus for characteristics signs of AD or death. Surviving mice were kept in isolation for 60 days post-inoculation with virulent virus and the mice were euthanised with chloroform and the trigeminal nerve ganglia collected in TEN.
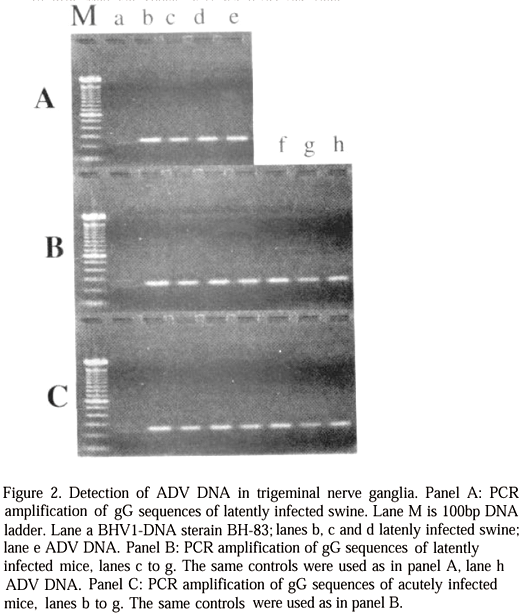
Homogenization of the trigeminal nerve ganglia was carried out by placing the tissue into a sterile 2.0ml glass homogenizer in 0.2ml of TEN. The tissue was homogenized completely using ten strokes of the plunger. Cell debris were pelleted by centrifugation at 2000´g for 10min. The supernatant was adjusted to 10% Nonidet P40, 1.0mg proteinase K /ml and incubated at 55ºC for 1-3 hours. The digestion tube was cooled off and 0.1ml KAc was added. After 30min, on ice, sediment was pelleted by centrifugation and the supernatant collected with ethanol. The precipitate was dissolved in 10mM Tris-HCl, pH 8.2; 1mM EDTA with 1mg/ml RNAse A. In order to assess the ADV status of a given test sample the use of PCR essentially as described by Flatschart & Resende (1998) was performed. Oligonucleotide primers, 22 base long, were chosen to amplify 260 base pairs (pb) sequence of the gene encoding the glycoprotein gG (Fig. 1). The primers were synthesized according to the sequence of the gene gG as previously reported (Rea et al., 1985).
The positive control for each test was the purified ADV DNA and negative controls were BHV-1 strain BH 83 DNA and trigeminal nerve ganglia DNA of uninfected mice.
Purified ADV DNA (1mg) was digested to completion with 50 units of Bam H I under the conditions recommended by the manufacturer ( Life Technologies, Inc.). The DNA fragments were separated on 20´20 cm horizontal 0.7% agarose slab gels and the Bam H I 7 fragment (Ladin et al., 1982) excised from the gel and purified using "Glassmax DNA isolation matrix system"(Life Technologies, Inc.), (Fig. 1). The purified Bam H I 7 fragment was labeled with 32P-dCTP by nick translation using a commercial kit (Life Technologies, Inc.). In a second alternative, another probe was prepared by digesting the 260bp PCR amplification product of gene gG with Bam H I to produce a 196 by internal fragment lacking the primer sequences. This fragment was labeled with biotin-7-dATP by nick translation according to manufacturers directions(Life Technologies, Inc.).
Prior to binding to nylon membrane (Hybond N+) (Amersham Corp.) or nitrocellulose membranes (Life Technologies, Inc.) samples of DNA were denatured by adding 0.1 volumes of 3M NaOH for 10min and then were chilled on ice. Denatured DNA samples were diluted in 0.125 X SSC containing 0.125N NaOH. The nylon membrane and filter pad were soaked in 0.4M Tris-HCl, pH 7.0 for 30min and placed in the dot-blot apparatus (Amersham Corp.). The denatured DNA samples were applied to the nylon membrane or nitrocellulose membranes and after 30min, gentle vacuum was applied. Finally, the DNA was fixed to the membrane by UV irradiation and allowed to dry at room temperature.
Dot blots to be hybridized to
32P-labeled probes were pre-hybridized in 3 X SSC, 0.1% (w/v) SDS, 10 X Denharts solution (2% w/v each of polyvinylpyrolidone, bovine serum albumin, ficoll) and 0.2M phosphate buffer, pH 6.8 for 6 hours at 65ºC. Hybridization was performed in pre-hybridization solution containing 0.1mg of denatured salmon sperm DNA/ml for 16 hours at 65ºC. Blots were rinsed in 2 X SSC, 1% (w/v) SDS at room temperature and washed at 65ºC with constant agitation for 30min each in 2 X SSC/ 1% SDS, 0.5 X SSC/0.25% SDS and 0.125 X SSC/ 0.0625% SDS. The blots were exposed to X-ray film overnight.
Dot blots to be hybridized to biotin-labeled probes were pre-hybridized in 6 X SSC/ 0.5% SDS, 0,1% BSA 5 X Denharts solution, 100mg/ml denatured salmon sperm DNA for 3 hours at 56ºC. For hybridization, the pre-hybridization solution was discarded and replaced with fresh pre-hybridization solution, but salmon sperm DNA was substituted with 60ng biotin-labeled probe. The filter with dot-blot was incubated at 45ºC for 18 hours. Three washing steps, of 30minutes each, were carried out at 45ºC for 18 hours. The signals of the biotin label were detected according to the manufacturers instructions included in the labeling kit (Life Technologies, Inc.)
RESULTS
No signs of AD were seen in the infected pigs exposed to 2´105 TCID50 of the ASB Piau strain of ADV in each nostril. The rectal temperature was elevated by 1ºC for 3-4 days and also a mild transient anorexia for three days was observed. In group A mice no clinical signs were observed after exposure to virulent virus. All mice in group B were severely affected and died 6-7 days after receiving infective virus showing characteristic signs of AD infection. These signs included cycles of prostration and hyperexcitability, anorexia, ataxia, paresis, paraplegia and pruritus.
Amplification with primers LVC A1 and A2 yielded a product of approximately 260bp. This product was clearly visualized as a sharp band on agarose gels (Fig. 2A, 2B and 2C) on all trigeminal nerve ganglia collected in infected pigs 350 days after exposure, in group A mice three weeks after exposure, in group A mice three weeks after exposure to virulent virus and also in group B mice at the time of death.
By dot-blot hybridization, ADV nucleic acids were detected in 6/6 of acute infected mice (Fig. 3 A). Both probes were unable to detect nucleic acid sequences in trigeminal nerve ganglia of latent infected mice (group A) and pigs (Fig.3 B and 3 C).
No signal was observed in DNA negative control samples (strain BH 83 of BHV-1).
DISCUSSION
The PCR confirmed ADV infection in all pigs and groups A and B of mice. The PCR was able to detect latent ADV in the trigeminal nerve ganglia of all experimentally infected pigs and group A and B of mice. The selection of trigeminal nerve ganglia was based on a previous report suggesting that this tissue is the most frequently involved in latency of ADV (Rziha et al., 1984). Under the amplification conditions used, the test has sufficient susceptibility to detect latent infected animals. The data suggests PCR is a valuable tool in the study of the infection status of a single animal, independently if this animal has acute or latent infection. Previous work reports on the low or variable frequency of detecting latent ADV using other methods (Van Oirschot & Fraser, 1984).
The dot blot hybridization procedure, using the Bam H I 7 fragment labeled with 32P dCTP and the 196 gene gG PCR product lacking the primer sequences labeled with biotin has potential for diagnosing acute ADV infection. But the results showed the level of sensitivity of both probes was not sufficient to detect ADV sequences in latent infected tissues and therefore the probes are not a laboratory method reliable to examine the presence of latent ADV infections. Otherwise, in many situations probes can be more attractive than PCR. Probes like those produced can be an excellent tool for studying viral replication and viral pathogenesis and would be more convenient for screening large number of samples. It is possible to detect viral genome with the use of the infected cell lisate method instead of DNA extraction, making the procedure less time-consuming and more attractive. For diagnostic proposes of acute cases, probes would be advantageous over tests like virus isolation, serum neutralization, capture ELISA and immune-fluorescence assay.
Finally, the results of the present study indicate the potential usefulness of the probes produced as a means of detecting ADV infections only in acute infections.
BIBLIOGRAPHIC REFERENCES
BASKERVILLE, A., McFERRAN, J.B., DOW, C. Aujeszky's disease in pigs.
Vet. Bull., v.43, p.465-480, 1973.
CARNEIRO, V., CARDIM, W.H. A doença de Aujeszky em suínos no Brasil. Arq. Inst. Biol. São Paulo, v.18, p.243-252, 1947.
CARVALHO, R., RESENDE, M, FRANCO, G. Teste imunoenzimático (ELISA) indireto para a detecção de anticorpos contra a doença de Aujeszky (VDA) em suinos. Arq. Bras. Med. Vet. Zootec., v.46, p.101-111, 1994.
FLATSCHART, R.B., RESENDE, M. Acute and latent infection in mice with a virulent strain of Aujeszky's disease virus. Rev. Microbiol. 1998 (submetido à publicação).
KOJNOK, J. The role of carrier sows in the spreading of Aujeszky's disease to suckling pigs. Data on Aujeszky's virus carriership among fattening pigs. Acta Vet. Acad. Sci. Hung., v.15, p.283-295, 1965.
LADIN, B.F., IHARA, S., HAMPL, H. et al. Pathway of assembly of herpesvirus capsids: An analysis using DNA+ temperature-sensitive mutants of pseudorabies virus. Virology, v.116, p.544-561. 1982.
LENNETTE, E.H., SCHMIDT, N.J., Eds. Diagnostic procedures for viral, rickettsial and chlamydial infections. Washington, APHA, 1979. 1138p.
MAES, R.K., SPATZ, S.J., THACKER, B.J. Nonradioactive probes to detect acute and latent pseudorabies virus (Aujeszky's disease) infections. In: IPVS CONGRESS, 10th, 1988, Rio de Janeiro: Proceedings..., Rio de Janeiro: IPVS, 1988. p.169.
REA, T.J., TIMMINS, J.G., LONG, G.W. et al. Mapping and sequence of the gene for the pseudorabies virus glycoprotein which accumulates in the medium of infected cells. J. Virol. v.54. p.21-29. 1985.
RZIHA, H.J., METTENLEITER, T.C., OHLINGER V. et al. Herpesvirus (pseudorabies virus) latency in swine: occurrence and physical state of viral DNA in neural tissues. Virology, v.155, p.600-613, 1988.
RZIHA, H.J., METTENLAITER, T., WITTMANN, G. Occurrence of pseudorabies virus DNA in latently infected pigs. In: WITTMANN, R. M., GASKELL, R. M., RZIHA, H. J. (ed). Latent Herpes Virus Infections in Veterinary Medicine. Holand: Martinus Nijhoff, 1984. p.429-433.
VAN OIRSCHOT, J.T., GIELKENS, A.L.J. In vitro and in vivo reactivation of latent pseudorabies virus in pigs born to vaccinated sows. Am. J. Vet. Res., v.45, p.567-571, 1984.
WHITE, A.K., CIACCI-ZANELLA, J., GALEOTA, J. et al. Comparison of the abilities of serologic tests to detect pseudorabies-infected pigs during the latent phase of infection. Am. J. Vet. Res., v.57, p.608-611, 1996.
- BASKERVILLE, A., McFERRAN, J.B., DOW, C. Aujeszky's disease in pigs. Vet. Bull., v.43, p.465-480, 1973.
- CARNEIRO, V., CARDIM, W.H. A doença de Aujeszky em suínos no Brasil. Arq. Inst. Biol. Săo Paulo, v.18, p.243-252, 1947.
- CARVALHO, R., RESENDE, M, FRANCO, G. Teste imunoenzimático (ELISA) indireto para a detecçăo de anticorpos contra a doença de Aujeszky (VDA) em suinos. Arq. Bras. Med. Vet. Zootec, v.46, p.101-111, 1994.
- FLATSCHART, R.B., RESENDE, M. Acute and latent infection in mice with a virulent strain of Aujeszky's disease virus. Rev. Microbiol. 1998 (submetido ŕ publicaçăo).
- KOJNOK, J. The role of carrier sows in the spreading of Aujeszky's disease to suckling pigs. Data on Aujeszky's virus carriership among fattening pigs. Acta Vet. Acad. Sci. Hung., v.15, p.283-295, 1965.
- LADIN, B.F., IHARA, S., HAMPL, H. et al. Pathway of assembly of herpesvirus capsids: An analysis using DNA+ temperature-sensitive mutants of pseudorabies virus. Virology, v.116, p.544-561. 1982.
- LENNETTE, E.H., SCHMIDT, N.J., Eds. Diagnostic procedures for viral, rickettsial and chlamydial infections. Washington, APHA, 1979. 1138p.
- MAES, R.K., SPATZ, S.J., THACKER, B.J. Nonradioactive probes to detect acute and latent pseudorabies virus (Aujeszky's disease) infections. In: IPVS CONGRESS, 10th, 1988, Rio de Janeiro: Proceedings..., Rio de Janeiro: IPVS, 1988. p.169.
- REA, T.J., TIMMINS, J.G., LONG, G.W. et al. Mapping and sequence of the gene for the pseudorabies virus glycoprotein which accumulates in the medium of infected cells. J. Virol. v.54. p.21-29. 1985.
- RZIHA, H.J., METTENLEITER, T.C., OHLINGER V. et al. Herpesvirus (pseudorabies virus) latency in swine: occurrence and physical state of viral DNA in neural tissues. Virology, v.155, p.600-613, 1988.
- RZIHA, H.J., METTENLAITER, T., WITTMANN, G. Occurrence of pseudorabies virus DNA in latently infected pigs. In: WITTMANN, R. M., GASKELL, R. M., RZIHA, H. J. (ed). Latent Herpes Virus Infections in Veterinary Medicine Holand: Martinus Nijhoff, 1984. p.429-433.
- VAN OIRSCHOT, J.T., GIELKENS, A.L.J. In vitro and in vivo reactivation of latent pseudorabies virus in pigs born to vaccinated sows. Am. J. Vet. Res, v.45, p.567-571, 1984.
- WHITE, A.K., CIACCI-ZANELLA, J., GALEOTA, J. et al. Comparison of the abilities of serologic tests to detect pseudorabies-infected pigs during the latent phase of infection. Am. J. Vet. Res, v.57, p.608-611, 1996.
Publication Dates
-
Publication in this collection
17 Apr 2001 -
Date of issue
Aug 1999
History
-
Received
10 Mar 1999