Resumos
Foi realizado um estudo por meio de simulação para avaliar a eficiência e o poder dos modelos aleatórios na estimação da localização e dos componentes de variância relativos a dois QTLs presentes no mesmo cromossomo, posicionados no mesmo intervalo, em intervalos adjacentes ou não. Admitiram-se diferentes tamanhos e números de famílias de irmãos completos e variâncias dos QTLs, em característica com herdabilidade igual a 0,25. A estimação dos parâmetros foi obtida por meio do método da máxima verossimilhança, baseado no quadrado da diferença de pares de irmãos, sob mapeamento por intervalo. As proporções de genes idênticos por descendência (IBD) dos QTLs foram estimadas a partir da proporção IBD de dois marcadores flanqueadores. Os fatores que mais influenciaram as estimativas dos parâmetros foram a proporção da variância atribuída aos QTLs e o número e o tamanho das famílias. Com número suficiente de famílias e de indivíduos nas famílias e altas proporções de variância genética, o modelo aleatório pode detectar QTLs com alto poder, apresentando estimativas das posições com boa acurácia.
mapeamento por intervalo; marcador genético; método dos pares de irmãos; modelo aleatório; QTL
A study was carried out by simulation to evaluate the efficiency and robustness of the random model approach for estimation of the QTL location and variance components in an outbred population with full-sib family structure. Two QTL were positioned in the chromosome in the same interval, in adjacent and at no adjacent intervals. The population was created with different sizes and numbers of families and variances due to QTL in a trait with h² = 0.25. The estimations of QTL parameters (locations and variance components) were based on the sib-pair approach. The proportions of genes identical-by-descent (IBD) at the two QTL were estimated from the IBD at two flanking markers. The most important factors afeccting the estimates of QTL parameters and power of detection were the proportion of variance due to QTL, and the number and size of the full-sibs families. Given a sufficient number of families and high proportions of genetic variance due to QTL, the random model approach can detect a QTL with high power and provides accurate estimates of the QTL position if there are no two QTL in the same interval.
genetic marker; interval mapping; random model; sib-pair method; QTL
ZOOTECNIA E TECNOLOGIA E INSPEÇÃO DE PRODUTOS DE ORIGEM ANIMAL
Mapeamento de QTL em famílias de irmãos completos por meio de modelos aleatórios
Mapping QTL in full-sib families using random model approach
M.V.G.B SilvaI,III; M.L. MartinezI,III; R.A. TorresII,III; P.S. LopesII,III; R.F. EuclydesII,III; M.A. MachadoI; W. ArbexI
IEmbrapa Gado de Leite Rua Eugênio do Nascimento, 610 Dom Bosco 36038-330 - Juiz de Fora, MG
IIDepartamento de Zootecnia UFV
IIIBolsista do CNPq
RESUMO
Foi realizado um estudo por meio de simulação para avaliar a eficiência e o poder dos modelos aleatórios na estimação da localização e dos componentes de variância relativos a dois QTLs presentes no mesmo cromossomo, posicionados no mesmo intervalo, em intervalos adjacentes ou não. Admitiram-se diferentes tamanhos e números de famílias de irmãos completos e variâncias dos QTLs, em característica com herdabilidade igual a 0,25. A estimação dos parâmetros foi obtida por meio do método da máxima verossimilhança, baseado no quadrado da diferença de pares de irmãos, sob mapeamento por intervalo. As proporções de genes idênticos por descendência (IBD) dos QTLs foram estimadas a partir da proporção IBD de dois marcadores flanqueadores. Os fatores que mais influenciaram as estimativas dos parâmetros foram a proporção da variância atribuída aos QTLs e o número e o tamanho das famílias. Com número suficiente de famílias e de indivíduos nas famílias e altas proporções de variância genética, o modelo aleatório pode detectar QTLs com alto poder, apresentando estimativas das posições com boa acurácia.
Palavras-chave: mapeamento por intervalo, marcador genético, método dos pares de irmãos, modelo aleatório, QTL.
ABSTRACT
A study was carried out by simulation to evaluate the efficiency and robustness of the random model approach for estimation of the QTL location and variance components in an outbred population with full-sib family structure. Two QTL were positioned in the chromosome in the same interval, in adjacent and at no adjacent intervals. The population was created with different sizes and numbers of families and variances due to QTL in a trait with h2 = 0.25. The estimations of QTL parameters (locations and variance components) were based on the sib-pair approach. The proportions of genes identical-by-descent (IBD) at the two QTL were estimated from the IBD at two flanking markers. The most important factors afeccting the estimates of QTL parameters and power of detection were the proportion of variance due to QTL, and the number and size of the full-sibs families. Given a sufficient number of families and high proportions of genetic variance due to QTL, the random model approach can detect a QTL with high power and provides accurate estimates of the QTL position if there are no two QTL in the same interval.
Keywords: genetic marker, interval mapping, random model, sib-pair method, QTL.
INTRODUÇÃO
A utilização de modelos aleatórios foi proposta, inicialmente, para a análise de dados de pares de irmãos (Haseman e Elston, 1972) em estudos de genética humana, onde há predominância de pequenas famílias de irmãos completos. Esse método é baseado na regressão linear dos quadrados das diferenças fenotípicas entre dois irmãos, dentro de uma família, em função da proporção de genes idênticos por descendência (IBD) compartilhados por eles. Apesar de ser considerado robusto para diversas distribuições dos dados e independência do modelo genético do QTL, esse método mostrou-se limitado, pois o efeito genético atribuído ao QTL e à taxa de recombinação estavam confundidos.
Na tentativa de separar a variância devido ao QTL e ao parâmetro de ligação e, ainda, localizar o QTL em uma posição específica do cromossomo, Fulker e Cardon (1994) desenvolveram o procedimento de pares de irmãos usando o mapeamento por intervalo. Entretanto, apesar do maior poder estatístico, esse procedimento é baseado no método dos quadrados mínimos e não otimiza o uso de toda a informação contida nos dados, como as propriedades distribucionais, como ocorre com o uso do método da máxima verossimilhança (ML).
Segundo Martinez e Vukasinovic (2000), o primeiro método para estimar a variância genética atribuída à determinada região cromossômica, com base na ML, chamado método de pontos múltiplos de IBD, foi proposto por Goldgar (1990). Schork (1993) expandiu este método de modo a estimar simultaneamente a variância de múltiplos intervalos e o efeito de ambiente comum partilhado por dois irmãos no mesmo ambiente. Embora esses métodos utilizassem marcadores flanqueadores para definir o segmento cromossômico, eles não foram elaborados para o mapeamento por intervalo.
Assim, Xu e Atchley (1995) propuseram o procedimento de mapeamento por intervalo baseado no método da ML desenvolvido por Goldgar (1990), em que se supõe distribuição normal dos valores genotípicos do QTL e ajustamento do QTL e do efeito poligênico como efeitos aleatórios para a análise de famílias de irmãos completos.
Este trabalho teve o objetivo de avaliar a eficiência e o poder deste método na estimação da localização e dos componentes de variância relativos a dois QTLs posicionados no mesmo intervalo, em intervalos adjacentes ou não, com diferentes tamanhos e números de famílias de irmãos completos e variâncias dos QTLs, em característica com herdabilidade igual a 0,25.
MATERIAL E MÉTODOS
Um modelo aleatório pode ser escrito como:
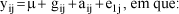
yij = valor observado da característica medido na jésima progênie da iésima família;
µ = constante comum a toda observação;
gij = efeito genético aditivo do QTL ~ N(0, sg2);
aij = efeito aditivo de todos os genes poligênicos, com exceção do QTL testado ~ N(0, sa2);
eij = efeito de ambiente ~ N(0, se2).
Assume-se que todos os efeitos aleatórios do modelo seguem distribuição normal; entretanto, se sa2e se2 são suficientemente grandes e normalmente distribuídos, não é exigida a normalidade para o efeito do QTL.
Para o uso desse modelo, pressupõe-se ausência de interferência e de dominância; os crossing-overs seguem distribuição de Poisson, ocorrem a uma distância de l Morgans, e são uniforme e independentemente distribuídos na região analisada.
A variância de yij, assumindo-se equilíbrio de ligação, é Var(yij) = s2 = sg2 + sa2 + se2 e a covariância entre dois irmãos completos não-endogâmicos, pertencentes à mesma família é Cov(yij, yij') = pqsg+½sa2, em que pq é a proporção de alelos IBD do suposto QTL compartilhada pelos irmãos.
O coeficiente associado à variância poligênica é ½ porque, em média, espera-se que dois irmãos completos compartilhem tais proporções de IBD. As proporções de IBD do QTL (p q) serão diferentes para cada par de irmãos, podendo variar de 0 a 1, para irmãos completos.
Com k irmãos completos na iésima família, a matriz de (co)variâncias entre os valores fenotípicos dos irmãos completos é

Com k irmãos em cada família, Ci é uma matriz k x k. O termo p q é a proporção de alelos IBD do QTL compartilhados pelos indivíduos j e j'. Na prática, entretanto, esta proporção não pode ser observada. Todavia, p q pode ser estimado por regressão linear, por meio da informação dos marcadores (Fulker e Cardon, 1994):
 , em que p1 e p2 são os valores IBD para os dois marcadores flanqueadores. Assim, substitui-se o termo p q por sua estimativa,
, em que p1 e p2 são os valores IBD para os dois marcadores flanqueadores. Assim, substitui-se o termo p q por sua estimativa,  , quando as variâncias são estimadas.
, quando as variâncias são estimadas.
Definindo-se  como a herdabilidade do suposto QTL,
como a herdabilidade do suposto QTL,  como a herdabilidade do componente poligênico e
como a herdabilidade do componente poligênico e  como a herdabilidade total, e admitindo-se distribuição normal multivariada dos dados, a função densidade conjunta das observações dentro das famílias é:
como a herdabilidade total, e admitindo-se distribuição normal multivariada dos dados, a função densidade conjunta das observações dentro das famílias é:

yi = [yi1, yi2, yi3, ..., yik]' é um vetor k x 1 de valores fenotípicos observados para k irmãos dentro da família i, e 1 = vetor k x 1 com todos os elementos iguais a 1.
O log da verossimilhança para n famílias de irmãos completos independentes é
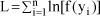
A função de verossimilhança relaciona a posição do QTL flanqueado pelos dois marcadores por meio de rik. Os parâmetros desconhecidos a serem estimados são µ, s2, hg2, ha2 e q1q. Para maximizar L no mapeamento por intervalo, considera-se a fração de recombinação entre o primeiro marcador e o suposto QTL (q1q) como uma constante e, gradualmente, vai-se aumentando seu valor e, conseqüentemente, diminuindo-se a distância entre o QTL e o marcador posicionado do lado direito (q2q), em todo o intervalo, repetindo-se o procedimento até que todo o genoma tenha sido percorrido. A estimativa ML da posição do QTL é determinada pelo valor de q1q que maximiza L e a hipótese de nulidade H0: hg2 = 0, ou seja, não existe QTL segregando no intervalo, pode ser testada por meio do teste de razão de verossimilhança (LR). A estimativa ML sob a hipótese de nulidade é denotada por L0. Assim, LR é dado por:
LR = -2(L0 L), que segue a distribuição de qui-quadrado com 2 ³ GL > 1 sob H0.
Informações genotípicas e fenotípicas para dois números de famílias (10 e 50 touros não-aparentados) foram geradas por meio da técnica de simulação de Monte Carlo. Foram geradas ainda famílias de irmãos completos de tamanhos diferentes (5 e 10 indivíduos), onde cada indivíduo possuía um segmento cromossômico de 60cM com quatro marcadores, igualmente distribuídos no cromossomo a intervalos de 20cM. Todos os marcadores possuíam seis alelos com a mesma freqüência. Dois QTLs, com quatro alelos codominantes com a mesma freqüência e efeitos aditivos, foram simulados em três situações:

As informações dos pais (touros e vacas) foram geradas alocando-se aleatoriamente os genótipos em cada loco, assumindo-se equilíbrio de Hardy-Weinberg. Admitiu-se serem desconhecidas as fases de ligação na geração parental. As informações das progênies foram geradas sob suposição de ausência de interferência; desse modo, um evento de recombinação em um intervalo não afeta a possibilidade de ocorrência de outra recombinação no intervalo adjacente. As freqüências de recombinação para cada loco foram calculadas usando-se a função de Haldane. Os dados fenotípicos foram gerados por simulação admitindo-se distribuição normal, média zero e variância igual a um, a partir do seguinte modelo:

yij = valor fenotípico do jésimo indivíduo na iésima família de irmãos completos;
µ = constante comum a toda observação;
qij = efeito dos genótipos dos QTLs do jésimo indivíduo;
si = contribuição do pai para o valor poligênico da progênie;
dij = contribuição da mãe para o valor poligênico da progênie;
fij= efeito da amostragem mendeliana;
eij = erro aleatório.
Para maior simplicidade não foram considerados efeitos fixos na simulação e admitiu-se que os touros não eram aparentados e que as vacas eram tomadas aleatoriamente da população e não possuíam parentesco entre si, ou com os touros.
De modo a testar o comportamento do modelo aleatório na detecção de QTL em gado de leite, foi escolhido o valor igual a 0,25 para a herdabilidade da característica e três diferentes proporções de variância explicada pelos QTLs (20, 40 e 100%), sendo a variância genética total obtida por meio da soma do componente dos QTLs e outro do efeito poligênico. O número de alelos dos QTLs foi igual a quatro e o dos marcadores, seis, sendo suas freqüências iguais.
Os dados foram analisados usando-se a máxima verossimilhança por meio da abordagem de mapeamento por intervalo, baseado no método de pares de irmãos proposto por Haseman e Elston (1972). A proporção de alelos IBD compartilhados por pares de irmãos relativa a cada loco do marcador foi calculada usando-se as equações fornecidas por Haseman e Elston (1972). A função de verossimilhança foi maximizada em relação aos parâmetros desconhecidos hg2, ha2 e s2 usando-se o algoritmo simplex (Nelder e Mead, 1965). O cromossomo de 60 cM foi percorrido a passos de 2 cM, entre um marcador e o subseqüente, da esquerda para a direita, calculando-se a razão de verossimilhança (LR) a cada passo, aceitando-se o maior valor de LR como a mais provável localização do QTL. Para cada combinação de parâmetros (tamanhos e números das famílias, proporção de variâncias do QTL etc.), a simulação e as análises foram repetidas cem vezes. As acurácias das estimações das posições dos QTLs foram determinadas usando o intervalo de confiança a 95%, estimado a partir da variância entre repetições e calculado como quatro vezes o erro-padrão empírico.
A distribuição empírica do teste LR foi gerada da mesma maneira para cada uma das combinações dos parâmetros sob a hipótese nula de que não havia nenhum QTL no segmento cromossômico. O nível de significância a = 0,05 foi escolhido para todas as análises. O valor empírico de limiar foi definido como o 95º percentil da distribuição empírica do teste LR sob H0. O poder de detecção foi definido como o percentual das repetições em que a hipótese nula foi rejeitada ao nível de significância de 5%
RESULTADOS E DISCUSSÃO
As médias e os respectivos intervalos de confiança das estimativas para as localizações dos QTLs, relativos a cem repetições, para as diferentes situações, proporções de variâncias genéticas devido aos QTLs e tamanhos da amostra podem ser observadas na Tab. 1.
Pode-se verificar que as estimativas das posições foram semelhantes para ambas proporções de variâncias estudadas (40 e 100%), para a situação onde os dois QTLs estavam presentes no mesmo intervalo (situação A). O mapeamento por intervalo não indicou corretamente os picos de LR no intervalo considerado, localizando um QTL entre os dois simulados, no meio do cromossomo (Fig. 1). Resultado semelhante foi obtido por Da et al. (2000), os quais verificaram que a utilização do mapeamento por intervalo pode identificar apenas um QTL em determinado intervalo que contenha dois QTLs. Nesse caso, a localização incorreta não terá reflexos importantes na seleção assistida por marcadores; todavia, poderá resultar em sérios problemas na identificação e no isolamento do(s) gene(s) associado(s) à característica quantitativa.
Em relação às duas variâncias estudadas nessa situação (40 e 100%), não houve diferenças entre elas, em relação a posições dos QTLs.
Quando os QTLs foram simulados em intervalos adjacentes (situação B), o mapeamento por intervalo mostrou dois picos (Fig. 2), relativos ao primeiro e ao segundo QTLs nas posições 15,0 e 22,9 cM, respectivamente. De modo geral, as estimativas de posição dos QTLs não foram próximas dos parâmetros verdadeiros, indicando que o mapeamento por intervalo, na presença de QTLs ligados em intervalos adjacentes, pode não ser o método apropriado na análise de famílias pouco numerosas. De fato, van der Beek et al. (1993) observaram baixas acurácias na estimação da posição de QTL ligados em famílias de irmãos completos compostas por dez indivíduos. Entretanto, segundo Goddard et al. (1999), aumento na acurácia para a detecção de QTLs em pequenas famílias de irmãos completos pode ser obtido se forem analisadas amostras selecionadas de irmãos, excluindo-se indivíduos não-informativos, concordando com as afirmações de Guo e Elston (2000). Tal afirmação é confirmada por Chatziplis e Haley (2000), os quais afirmaram que pares de irmãos completos, oriundos de famílias com baixa variância, não estariam segregando para o QTL e, dessa forma, são não-informativos para a análise.
A despeito do tamanho da amostra, Da et al. (2000) verificaram que a detecção de QTL, usando modelos de regressão no mapeamento por intervalo composto, também considerou erroneamente dois QTLs, presentes no mesmo intervalo, como se fossem um, sugerindo que a consideração do intervalo como contínuo ou discreto pode influenciar a estimação correta da posição dos QTLs.
Na situação B, quando a variância era menor (40%), a estimativa da localização foi a mesma para ambos os QTLs. Quando a variância genética foi integralmente atribuída aos QTLs, houve o aparecimento de dois picos, evidenciando a presença dos dois QTLs, embora com estimativas viesadas.
Para a situação C, onde os QTLs foram posicionados em intervalos não-adjacentes, pode ser observado na Tab. 1 que as estimativas foram se tornando menos viesadas com o aumento do número de informações dentro de famílias e a variação genética explicada pelos QTLs (20, 40 e 100%), resultado este também encontrado por Silva (2002), embora em nenhum dos casos a posição estimada tenha sido próxima da verdadeira. Observa-se ainda que, de modo geral, as posições estimadas dos QTLs tenderam a ficar sempre à direita dos QTLs simulados.
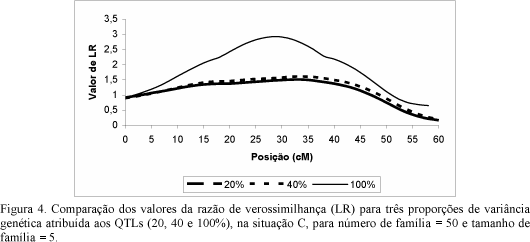
Nas famílias com menor número de indivíduos houve o aparecimento de apenas um pico no meio do cromossomo (Fig. 3 e 4), mais evidente quando se aumentou o número de famílias de 10 para 50. Nas populações nas quais foram analisados 10 indivíduos por família, houve tendência de aparecimento de dois picos (Fig. 5 e 6), que, apesar de não indicarem corretamente as posições, evidenciam que o número e o tamanho das famílias avaliadas têm influência na identificação mais correta de dois QTLs posicionados no mesmo cromossomo.
Segundo van der Beek et al. (1995), é necessário menor número de animais em experimentos envolvendo estruturas de famílias de irmãos completos em relação a experimentos envolvendo famílias de meios-irmãos. Silva (2002) verificou que as localizações com maior acurácia foram obtidas ao serem analisados dados referentes a 50 famílias com 40 indivíduos cada uma. Confrontando os resultados desse autor com os encontrados neste estudo, verificou-se que a maior amostra estudada (50 famílias com 10 indivíduos cada uma) foi insuficiente para localizar com alta acurácia e precisão os QTLs, sugerindo que o número razoável de indivíduos por família, para as variâncias e herdabilidade estudadas, estaria entre 10 e 40. Dessa forma, pode ser interessante a simulação de situações para a comparação da estimação da localização e do poder de detecção de poucas famílias de meios-irmãos, compostas por numerosos indivíduos, com muitas famílias de irmãos completos, compostas por poucos indivíduos.
Do ponto de vista prático, essas simulações poderiam auxiliar no delineamento de experimentos, pois em virtude das limitações na capacidade reprodutiva de fêmeas, as famílias de irmãos completos são em geral menores que aquelas compostas por meios-irmãos, o que é particularmente verdadeiro em bovinos e suínos.
Considerando-se as situações A, B e C, as estimativas mais viesadas foram obtidas quando os QTLs estavam posicionados nas extremidades do cromossomo, semelhante aos resultados obtidos por Mangin et al. (1999). Ressalte-se, ainda, que ocorreram reduções dos intervalos de confiança com os aumentos da variância explicada pelo QTL e do tamanho da amostra.
O poder de detecção empírico, definido como o percentual de repetições nas quais o valor máximo de LR ultrapassou o valor de limiar empírico obtido pela simulação de dados sob H0, é apresentado na Tab. 2. O poder de detecção foi altamente dependente da proporção da variância genética, do número de famílias, do tamanho destas famílias e das posições dos QTLs. Independentemente da variância atribuída aos QTLs, o poder de detecção foi igual a zero para as situações e combinações de parâmetros cujo número de famílias ou de indivíduos era cinco, mesmo resultado obtido para variâncias dos QTLs iguais a 20 e 40%.
Nas situações A e B, nas quais havia 50 famílias com 10 indivíduos, o poder de detecção, apesar de superior a zero, foi baixo, indicando que as posições dos QTLs no cromossomo estão relacionadas com o poder de detecção. Ainda, ao se distribuir a variância genética para dois QTLs, talvez ocorra alguma perda no poder de detecção, quando comparado às situações em que somente um suposto QTL seja identificado, como nos trabalhos de van der Beek (1996) e Martinez et al. (1999). Nesse caso, talvez maior número de repetições seja necessário para estimar o poder com maior precisão e assegurar que os resultados não reflitam flutuações amostrais.
Outro ponto a ser destacado é que se admitiu que os marcadores possuíam freqüências iguais na população. Provavelmente, o poder estatístico seria reduzido caso as freqüências fossem diferentes, em razão da influência dos QTLs sobre a média da característica, pois um dos genótipos poderia ter maior freqüência e, desse modo, tornaria assimétrica a distribuição normal da característica.
O poder para a detecção foi altamente dependente do tamanho da amostra, das posições e da proporção da variância genética explicada pelos QTLs, pois somente quando havia 50 famílias de 10 indivíduos cada uma, QTLs posicionados em intervalos não-adjacentes e toda a variação explicada por eles, o poder foi próximo ao considerado como satisfatório (70%). A dependência da proporção da variância foi também observada por Martinez et al. (1999), enquanto outros estudos, como o realizado por Blackwelder e Elston (1982), mostraram que o tamanho da amostra e o número de alelos do marcador são também fatores importantes e que contribuem para o aumento do poder de detecção. Elevações no poder de detecção com o aumento do número de famílias de irmãos completos e de progênies por família também foram obtidas por van der Beek (1996).
CONCLUSÕES
O mapeamento por intervalo, baseado em modelos aleatórios, pode ser aplicado em populações de irmãos completos para a identificação de QTLs responsáveis por grande proporção da variação genética são detectados com alto poder, principalmente em populações compostas por, no mínimo, 50 famílias de irmãos completos, com mais de 10 indivíduos cada uma. O método não localiza com eficiência QTLs de pequenos efeitos, principalmente se estiverem localizados próximos às extremidades do cromossomo. Por ser um método simples e robusto, o modelo aleatório pode ser recomendado para a varredura rápida do genoma, aplicando-se, em seguida, análises mais refinadas nos intervalos em que for indicada a existência de um suposto QTL. O modelo aleatório não consegue separar com eficiência as variâncias poligênicas e dos QTLs. Métodos mais refinados devem ser usados para estimar a variância dos QTLs.
Recebido para publicação em 21 de julho de 2003
Recebido para publicação, após modificações, em 3 de novembro de 2003
E-mail: marcos@cnpgl.embrapa.br.
- BLACKWELDER, W.C.; ELSTON, R.C. Power and robustness of sib-pair linkage tests and extension to larger sibships. Comum. Statist. Theor. Methods, v.11, p.449-484, 1982.
- CHATZIPLIS, D.; HALEY, C.S. Selective genotyping for QTL detection using sib pair analysis in outbred populations with hierarchical structures. Genet. Sel. Evol, v.32, p.547-560, 2000.
- DA, Y.; van RADEN, P.M.; SCHOOK, L.B. Detection and parameter estimation for quantitative trait loci using regression models and multiple markers. Genet. Sel. Evol., v.32, p.357-381, 2000.
- FULKER, D.W.; CARDON, L.R. A sib-pair approach to interval mapping of quantitative trait loci. Am. J. Hum. Genet., v.47, p.957-967, 1994.
- GODDARD, K.A.B.; GOODE, E.L.; ROZEK, L.S. et al. Impact of structure on the power of linkage tests using sib-pair methods. Genet. Epidemiol., v.17, Suppl.1, p.S575-S579, 1999.
- GOLDGAR, D.E. Multipoint analysis of human quantitative genetic variation. Am. J. Hum. Genet., v.47, p.957-967, 1990.
- GUO, X.; ELSTON, R.C. Two-stage global search designs for linkage analysis II: Including discordant relative pairs in the study. Genet. Epidemiol., v.18, p.111-127, 2000.
- HASEMAN, J.K.; ELSTON, R.C. The investigation of linkage between a quantitative trait and a marker locus. Behav. Genet., v.2, p.3-19, 1972.
- MANGIN, B.; GOFFINET, B.; LE ROY, P. et al. Alternative models for QTL detection in livestock. II. Likelihood approximations and sire marker genotype estimations. Genet. Sel. Evol., v.31, p.225-237, 1999.
- MARTINEZ, M.L.; VUKASINOVIC, N. Algoritmo para cálculo da proporção de gens idênticos por descendência, para mapear QTL em famílias de meio-irmãos. Rev. Bras. Zootec., v.29, p.443-451, 2000.
- MARTINEZ, M.L.; VUKASINOVIC, N.; FREEMAN, A. E. Random model approach for QTL mapping in half-sib families. Genet. Sel. Evol., v.31, p.319-340, 1999.
- NEIMANN-SORENSON, A.; ROBERTSON, A. The association between blood groups and several production characteristics in three Danish cattle breeds. Acta Agric. Scand, v.11, p.163-196, 1960.
- NELDER, J.A.; MEAD, R. A simplex method for function minimization. Comput. J., v.7, p.308-313, 1965.
- SCHORK, N.J. Extended multipoint identy-by-descendent analysis of human quantitative traits: efficiency, power, and modeling considerations. Am. J. Hum. Genet., v.53, p.1306-1393, 1993.
- SILVA, M.V.G.B. Utilização de modelos aleatórios na estimação da localização de QTL em famílias de meios-irmãos. 2002. 112f. Tese( Doutorado em Genética e Melhoramento). Universidade Federal de Viçosa, Viçosa, MG.
- van der BEEK, S. The use of genetic markers in poultry breeding, 1996. 175f. Thesis (PhD in Animal Breeding)- Wageningen University, Wageningen, Netherlands.
- van der BEEK, S.; GRÖEN, A.F.; van ARENDONK, J.A.M. Evaluation of designs for reference families for livestock linkage mapping experiments. Anim. Biotech., v.4, p.163-182, 1993.
- van der BEEK, S.; van ARENDONK, J.A.M.; GRÖEN, A.F. Power of two- and three-generation QTL mapping experiments in na outbred population containing full-sib or half-sib families. Theor. Appl. Genet., v.91, p.1115-1124, 1995.
- XU, S.; ATCHLEY, W.R. A random model approach to interval mapping of quantitative trait loci. Genetics, v.141, p.1189-1197, 1995.
Datas de Publicação
-
Publicação nesta coleção
17 Jun 2004 -
Data do Fascículo
Abr 2004
Histórico
-
Recebido
21 Jul 2003