Resumos
Estudou-se a filogenia do gene da miogenina, um membro da família MyoD, reguladora da miogênese, que ocorre durante o desenvolvimento embrionário, e sua história evolutiva em espécies domésticas que apresentem seqüências de DNA depositadas no Genbank, comparando-se o índice de substituição de nucleotídeos não-sinônimos pelo índice de substituição sinônima. Valores maiores do que um (1) indicaram que o gene sofreu mudanças que tornaram o organismo mais adaptado ao ambiente. As árvores filogenéticas foram obtidas por máxima verossimilhança, e os índices de substituição sinônima e não-sinônima foram analisadas pelo método de parcimônia. Os resultados indicaram que, provavelmente, o gene sofreu evolução adaptativa no grupo Ruminantia, Bos taurus e Ovis aries, depois que essas espécies divergiram do ancestral comum. Para as outras espécies analisadas, o gene parece ter evoluído de modo conservativo.
miogênese; seqüenciamento de DNA; PCR
The myogenin gene, a member of the MyoD gene family, is a regulator of the myogenesis that takes place during the embryonic development. The objective of this study was to perform a phylogenic analysis of the myogenin gene to study its evolutionary history in the domestic species that have the sequencing data deposited in the Genbank. One common method to detect a gene evolution is made by comparing the ratio of nonsynonymous nucleotide substitution by the ratio of synonymous substitutions. Values greater than one (1) means that the gene has gone through changes that made the organism more adapted to the environment. The phylogenetic trees were obtained by maximum likelihood and the synonymous and nonsynonymous substitution rates were analyzed by the parsimony method. The results point out that probably the gene suffered an adaptive evolution in the Ruminantia group, Bos Taurus and Ovis aries, after these species diverged from their common ancestral. In the other species, the gene seems to be evolved in a conservative way.
myogenesis; DNA sequencing; PCR
ZOOTECNIA E TECNOLOGIA E INSPEÇÃO DE PRODUTOS DE ORIGEM ANIMAL
Análise filogenética do gene da miogenina
Phylogenetic analysis of the myogenin gene
A.S. SchierholtI; I. FonsecaI; P.V. SilvaI; S.R. PaivaII; L.C.S. ChavesI; P.S. LopesI; D.A. FariaI; S.E.F. GuimarãesI, * * Autor para correspondência ( Corresponding author) Av. P.H. Holfs, s/n 36571-000 Viçosa, MG E-mail: sfacioni@ufv.br
IUniversidade Federal de Viçosa
IIEmbrapa Recursos Genéticos e Biotecnologia Brasília, DF
RESUMO
Estudou-se a filogenia do gene da miogenina, um membro da família MyoD, reguladora da miogênese, que ocorre durante o desenvolvimento embrionário, e sua história evolutiva em espécies domésticas que apresentem seqüências de DNA depositadas no Genbank, comparando-se o índice de substituição de nucleotídeos não-sinônimos pelo índice de substituição sinônima. Valores maiores do que um (1) indicaram que o gene sofreu mudanças que tornaram o organismo mais adaptado ao ambiente. As árvores filogenéticas foram obtidas por máxima verossimilhança, e os índices de substituição sinônima e não-sinônima foram analisadas pelo método de parcimônia. Os resultados indicaram que, provavelmente, o gene sofreu evolução adaptativa no grupo Ruminantia, Bos taurus e Ovis aries, depois que essas espécies divergiram do ancestral comum. Para as outras espécies analisadas, o gene parece ter evoluído de modo conservativo.
Palavras-chave: miogênese, seqüenciamento de DNA, PCR
ABSTRACT
The myogenin gene, a member of the MyoD gene family, is a regulator of the myogenesis that takes place during the embryonic development. The objective of this study was to perform a phylogenic analysis of the myogenin gene to study its evolutionary history in the domestic species that have the sequencing data deposited in the Genbank. One common method to detect a gene evolution is made by comparing the ratio of nonsynonymous nucleotide substitution by the ratio of synonymous substitutions. Values greater than one (1) means that the gene has gone through changes that made the organism more adapted to the environment. The phylogenetic trees were obtained by maximum likelihood and the synonymous and nonsynonymous substitution rates were analyzed by the parsimony method. The results point out that probably the gene suffered an adaptive evolution in the Ruminantia group, Bos Taurus and Ovis aries, after these species diverged from their common ancestral. In the other species, the gene seems to be evolved in a conservative way.
Keywords: myogenesis, DNA sequencing, PCR
INTRODUÇÃO
Genes que desempenham algum tipo de papel importante sobre características que influenciem na sobrevivência do animal, ou genes que otimizem a eficiência de uso de energia, de maneira que o saldo de energia disponível para a reprodução seja maior, são mais prováveis de terem sido submetidos à pressão de seleção positiva durante a sua história evolutiva. Um método comum para se detectar a evolução de um gene é por meio da comparação da taxa de substituição de nucleotídeos não-sinônima (Ka) pela taxa de substituição sinônima (Ks) entre as espécies. Muitos métodos estão disponíveis para calcular a razão Ka/Ks, desde o simples método de Nei e Gojobori (1986) até o computacionalmente mais intensivo e altamente parametrizado de máxima verossimilhança de Yang e Nielsen (2000). O método PBL de Pamilo, Bianchi e Li citado por Liberles et al. (2001), um dos mais populares e de bom desempenho, foi modificado por Benner et al. (1998) e chamado de PBLSB, usando implementação de reconstruções de seqüências ancestrais. Essa modificação permitiu a comparação de taxas de Ka/Ks ao longo dos ramos de uma árvore filogenética ao invés de comparação simples das seqüências par a par (Liberles et al., 2001).
A massa muscular animal é uma característica importante para a sobrevivência do animal e, por isso, modificações que ocorram nos genes envolvidos na miogênese podem oferecer aos animais melhor eficiência na utilização desse tecido (Tellgren et al., 2004).
O processo de miogênese pode ser dividido em duas fases: determinação e diferenciação. A determinação é o evento em que as células pluripotentes que estão se multiplicando são mobilizadas para o processo miogênico, transformando-se em mioblastos. A diferenciação ocorre quando os genes dos miotúbulos iniciam sua expressão músculo-específica, então os mioblastos param de se multiplicar, e as fibras musculares passam a crescer quase que somente em volume (Coutinho et al., 1999). A miogenina tem papel importante durante a miogênese, pois sua expressão marca o início da diferenciação, conseqüentemente, quando se expressa a miogenina, as fibras musculares desenvolvem-se a partir dos mioblastos previamente formados.
Os objetivos desse trabalho foram estudar a evolução do gene da miogenina dentro das algumas espécies de animais domésticos que possuem o gene seqüenciado e verificar se o gene sofreu pressão de seleção positiva em algum período, durante a evolução das espécies.
MATERIAL E MÉTODOS
As seqüências do gene da miogenina das espécies usadas para as análises filogenéticas foram obtidas do Genbank1 1 Genbank http://www.ncbi.nlm.nih.gov. , sendo os números de acesso para cada espécie escolhidos com base na qualidade das informações depositadas.
No caso de Ovis aries, apenas um seqüenciamento havia sido depositado, e este não estava completo, não contendo o início e o final do gene. Na análise, também foi considerada a espécie Sus scrofa, usando uma seqüência consenso, obtida a partir do seqüenciamento de todo o gene da miogenina em raças comerciais e na raça nativa brasileira Piau. As espécies e o número de acessos analisados estão listados na Tab. 1.
As análises foram feitas considerando as seqüências (caracteres) do fragmento que se refere à região do mRNA maduro a ser traduzido, ou seja, a união do éxon 1, do éxon 2 e do éxon 3. Foram comparados, separadamente, os fragmentos que compõem o domínio básico e o domínio helix-loop-helix (HLH), que são os dois domínios funcionais encontrados no éxon 1.
As regiões estudadas obtidas do Genbank foram editadas a partir do programa Basic Local Alignment Search Tool (BLAST); Altschul et al. (1997), usando como referência as seqüências obtidas no seqüenciamento da unidade toxonômica operacional (OTU) Sus scrofa. O tamanho dos domínios básico e HLH foi também obtido no Genbank. As seqüências dos fragmentos de todas as OTUs foram alinhadas com o programa Clustal X (Thompson et al., 1997).
Os alinhamentos foram analisados utilizando-se o programa Model Test 3.5, desenvolvido por Posada e Crandall (1998), que usa a razão de máxima verossimilhança para determinar, com base nas características dos seqüenciamentos alinhados, quais parâmetros serão usados na construção da árvore filogenética e as estimativas de tais parâmetros.
O modelo usado para a construção da árvore filogenética deve ser capaz de incorporar heterogeneidade nas taxas de substituição nucleotídica ao longo dos sítios, pois, segundo Uzzel e Corbin (1971), nem todos os sítios evoluem na mesma taxa de substituição, como, por exemplo, aqueles sob forte restrição seletiva. A distribuição mais usada para modelagem é chamada de função gama.
Para a construção das árvores filogenéticas, foi utilizado o programa PAUP 4.0 beta 1.0 (Swofford, 1999), usando os arquivos criados pelo alinhamento com o Clustal X. A topologia das árvores foi construída empregando o método de máxima verossimilhança, com os modelos evolutivos selecionados e os parâmetros estimados com a utilização do programa Model Test 3.5. Para cada um dos fragmentos estudados, foi escolhido um modelo evolutivo de máxima verossimilhança, baseado nas características das seqüências de nucleotídeos desses fragmentos. As árvores foram enraizadas usando o grupo externo de Gallus gallus e Meleagris gallopavo.
O cálculo da taxa Ka/Ks foi realizado empregando o método PBLSB (Benner et al., 1998), segundo as fórmulas:
Ka = (L2 * B2 + L0 * K0) / (2 * L2 / 3 + L0) e
Ks = (L2 * A2 + L4 * A4) / (L2 + L4) + B4, em que:
Ka = taxa de transição não-sinônima; Ks = taxa de transição sinônima; L0 = sítio não degenerado; L2 = sítio duas vezes degenerado; L4 = sítio quatro vezes degenerado, respectivamente; Ki = Ai + Bi; Ai e Bi = taxa de transição e transversão para o i-ésimo tipo de sítio, respectivamente (i = 0, não degenerado; 2, duas vezes degenerado; e 4, quatro vezes degenerado).
As seqüências dos fragmentos analisados, alinhadas pelo Clustal X, foram usadas para a reconstrução dos estados ancestrais. Como o algoritmo trabalha com códons, foi realizado um ajuste em que todo códon, no qual houvesse um gap (espaço), fosse preenchido por gaps nas seqüências alinhadas.
Esse procedimento deve ser tomado porque o algoritmo que estima Ka/Ks, da maneira que é implementado na ferramenta do cálculo, não lida com frameshifts (mudanças na fase de leitura do gene), pois com muita freqüência tais frameshifts são apenas erros no arquivo de entrada. O programa simplesmente não leva em consideração tais casos diretamente, pois as seqüências com frameshift terão uma taxa muito alta de substituições não-sinônimas e irão, provavelmente, gerar um alto valor de Ka/Ks, se a porção que sofreu frameshift for de comprimento significativo.
RESULTADOS E DISCUSSÃO
Três conjuntos de dados, um com base na seqüência completa que codifica o mRNA do gene e outros dois, baseados nas seqüências que codificam os domínios funcionais HLH e básico, presentes no éxon 1, foram usados para gerar três árvores filogenéticas por meio de máxima verossimilhança para estas seqüências. Procedeu-se à análise dos domínios básico e HLH, separadamente, em razão de o primeiro ser o resíduo da proteína responsável pela interação com o DNA, e o segundo ser a ligação entre os monômeros de bHLH para formarem dímeros funcionais. Há grande chance de que as alterações nesses fragmentos possam levar à modificação na eficiência do funcionamento da proteína.
Essa análise diferenciada é aconselhada quando há informações prévias que permitam hierarquizar os sítios em classes, em que se esperam existir diferentes pressões seletivas e, dessa maneira, diferentes taxas Ka/Ks (Yang e Nielsen, 2002).
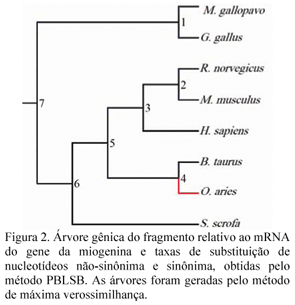
Na Fig. 1 está representado o alinhamento das seqüências do domínio HLH, usado na construção da árvore gênica desse fragmento do gene da miogenina.
Com o alinhamento das seqüências, podem-se avaliar quais parâmetros devem ser considerados na construção da árvore filogenética para cada fragmento. Os parâmetros para cada fragmento estudado foram estimados utilizando-se o programa Model Test 3.5, permitindo selecionar os seguintes modelos: para o fragmento de mRNA, o modelo evolutivo que melhor descreveu a heterogeneidade nas taxas de substituição dos caracteres foi o HKY85 + G, considerando que os sítios de substituição possuem distribuição gama, que a freqüência dos quatro nucleotídeos nem sempre é similar e que substituições do tipo transição ocorrem mais freqüentemente que as do tipo transversão; para os fragmentos domínio básico e domínio HLH, o modelo selecionado foi o K80+G, considerando que as substituições do tipo transição são mais freqüentes que as do tipo transversão e que os sítios de substituição apresentam distribuição gama.
Normalmente, os evolucionistas estão interessados na árvore filogenética que representa a história evolutiva de um grupo de espécies. Entretanto, quando a árvore é construída apenas com um gene, ela pode não representar estritamente a história evolutiva da espécie, mas sim a do gene, chamada de árvore gênica. Nesse caso, todas as três árvores mostraram exatamente a mesma topologia, como pode ser observado nas Fig. 2, 3 e 4, com o grupo externo formado pelas OTUs, G. gallus e M. gallopavo, como foi pré-estabelecido.
Como pode ser observado na árvore gênica, as OTUs mais proximamente relacionadas são B. taurus e O. aries, formando o grupo monofilético Ruminantia, e M. musculus e R. norvegicus, formando o grupo monofilético Rodentia. Essa árvore gênica não representa a história evolutiva das espécies, pois apresenta a OTU S. scofa formando um grupo monofilético com Rodentia. A árvore filogenética, representando a história evolutiva contendo essas OTUs, apresentaria o S. scrofa e Ruminantia formando um grupo monofilético chamado Artiodactila.
Com a árvore gênica pronta, a evolução das seqüências foi analisada para o gene da miogenina, a partir do método PBLSB (Benner et al., 1998), para a reconstrução dos estados dos caracteres ancestrais, juntamente com um método para contagem de sítios de substituições sinônimas e não-sinônimas.
Nas Tab. 2, 3 e 4 são mostrados os valores de Ka, Ks e da razão Ka/Ks obtidos nos ramos das árvores filogenéticas para cada fragmento estudado. Os nós e os ramos pertencentes a cada valor das tabelas podem ser visualizados, respectivamente, nas Fig. 2, 3 e 4. Primeiramente, pode-se notar o nível de conservação que o gene apresenta, e em todos os fragmentos estudados parece estar sob pressão de seleção positiva o ramo que leva à espécie O. áries (Tab. 2, 3 e 4 e Fig. 2, 3, e 4), exatamente a OTU que não apresentava a seqüência completa.
No fragmento referente ao mRNA, todos os ramos, a não ser os do grupo Ruminantia (ramos do nó 4, Tab. 2), apresentaram um valor de Ka/Ks menor que 0,3, e a grande maioria menores que 0,1. Isso indica que a proteína gerada pelo gene da miogenina, selecionada por milhões de anos, atingiu sua função otimizada previamente à divergência do grupo interno e do grupo externo. Isso implica uma pressão de seleção negativa ou evolução conservativa, em a que maioria das mutações não-sinônimas era prejudicial à adaptação das espécies. Dentro do grupo Ruminantia, o ramo que leva à OTU Bos taurus, o valor foi de 0,37 (ramo 1 do nó 4, Tab. 2) e no que leva à Ovis áries, de 2,42 (ramo 2 do nó 4, Tab. 2).
A mesma conclusão pode ser tirada para os outros dois fragmentos. Com relação ao domínio básico, novamente o ramo que leva à OTU O. aries se mostrou sob pressão de seleção positiva, cujo valor da razão Ka/Ks foi de 1,68 (ramo 2 do nó 2, Tab 3). No domínio HLH, a seleção apresentou-se muito mais conservativa, e quase todos os ramos apresentaram razão Ka/Ks igual a zero, com exceção dos ramos que levam às OTUs O. aries e B.taurus, que apresentaram razões elevadas de 13,15 e 19,57, respectivamente (Tab 3). Tais taxas indicam que as espécies analisadas do grupo Ruminantia estiveram sob grande pressão de seleção positiva.
A quantidade de informações relativas ao gene da miogenina ainda é muito escassa em banco de dados, como o Genbank, com poucas espécies com o gene seqüenciado. Isso prejudica a realização de um delineamento metodológico mais desejável, que inclua outras raças de suínos selvagens ou parentes próximos, como o javali. Espécies com informações muito distantes umas das outras podem gerar uma perda na qualidade da reconstrução dos estados ancestrais, especialmente quando o tamanho do ramo sob estudo aumenta, pois isso leva à diluição de um episódio de adaptação positiva (Ka/Ks > 1), com episódios de evolução conservativa.
Apesar da distância entre as OTUs, é importante ressaltar que a evolução do gene da miogenina ocorre em uma dinâmica diferente da evolução das espécies. A maneira conservativa que o gene evolui faz com que as espécies acabem sendo proximamente relacionadas quando se considera a seqüência de nucleotídeos do gene da miogenina, ou seja, os caracteres que forneceram as informações para a construção das árvores gênicas. Além disso, métodos baseados em parcimônia podem superar os métodos baseados em máxima verossimilhança na análise de seqüências proximamente relacionadas e são conhecidos por subestimar a quantidade de substituições em comparações mais distantemente relacionadas (Yang e Nielsen, 2002).
Esses resultados devem ser testados e confirmados com outros testes, pois a reconstrução das seqüências de ancestrais nem sempre é acurada, até mesmo quando são empregados critérios de máxima parcimônia, que usem delineamento mais apropriado para esse tipo de estudo e que permitam analisar com mais precisão a história evolutiva do gene da miogenina.
Com exceção do ramo que leva ao grupo Ruminantia, o gene da miogenina evolui sob seleção conservativa pelo menos desde a divergência dos mamíferos das aves. Sobre a evolução do gene no ramo que leva às espécies O. aries e B. taurus, o gene sofreu polimorfismos no domínio HLH, que possivelmente promoveram maior adaptabilidade desses organismos a seus ambientes. Os resultados constatam que é pequena a possibilidade de encontrar variantes no gene da miogenina para usá-lo em programas de melhoramento de suínos
AGRADECIMENTOS
Apoio financeiro CAPES, CNPq, FAPEMIG e FINEP.
Recebido em 16 de outubro de 2006
Aceito em 13 de dezembro de 2007
- ALTSCHUL, S.F.; MADDEN, T.L.; SCHAFFER, A.A. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Ac. Res., v.25, p.3389-3402, 1997.
- BENNER, S.A.; TRABESINGER, N.; SCHREIBER, D. Post genomic science: converting primary structure into physiological function. Adv. Enzime Regul., v.38, p.155-180, 1998.
- COUTINHO, L.L.; GABRIEL, J.E.; ALVARES, L.E. Controle molecular do desenvolvimento da musculatura esquelética. In: SIMPÓSIO INTERNACIONAL DE GENÉTICA E MELHORAMENTO ANIMAL, 1999, Viçosa. Anais.. Viçosa, 1999. p.355-376.
- LIBERLES, D.A.; SCHREIBER, D.R.; GOVINDARAJAN, S. The adaptive evolution database (TAED). Genome Biol., v.2, p.0028.1-0028.6, 2001.
- NEI, M.; GOJOBORI, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol, v.3, p.418-426, 1986.
- POSADA, D.; CRANDALL, K.A. Model test: testing the model of DNA substitution. Bioinformation, v.14, p.817-818, 1998.
- SWOFFORD, D.L. (Ed). PAUP: Phylogenetic analysis using parsimony and others methods Versão 4.0. Sunderland: Sinauer Associates Inc., 1999. 213p.
- TELLGREN, Å.; BERGLUND, A.C.; SAVOLAINEN, P. et al. Myostatin rapid sequence evolution in ruminants predates domestication. Mol. Phyl. Evol, v.33 p.782-790, 2004.
- THOMPSON, J.D.; GIBSON, T.J.; PLEWNIAK. F. et al. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Ac. Res., v.24, p.4876-4882, 1997.
- UZZEL. T.; CORBIN, K.W. Fitting discrete probability distribution to evolutionary events. Science, v.172, p.1089-1096, 1971.
- YANG, Z.; NIELSEN, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol, v.17, p.32-43, 2000.
- YANG, Z.; NIELSEN, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol, v.19, p.908-917, 2002.
Datas de Publicação
-
Publicação nesta coleção
05 Maio 2008 -
Data do Fascículo
Fev 2008
Histórico
-
Aceito
13 Dez 2007 -
Recebido
16 Out 2006